Aducanumab for the Treatment of Alzheimer’s Disease
RELEASE DATE
October 1, 2021
EXPIRATION DATE
October 31, 2023
FACULTY
G. Blair Sarbacker, PharmD, FAPhA
Professor of Pharmacy Practice
Presbyterian College School of Pharmacy
Clinton, South Carolina
FACULTY DISCLOSURE STATEMENTS
Dr. Sarbacker has no potential or actual conflicts of interest in relation to this activity.
Postgraduate Healthcare Education, LLC does not view the existence of relationships as an implication of bias or that the value of the material is decreased. The content of the activity was planned to be balanced, objective, and scientifically rigorous. Occasionally, authors may express opinions that represent their own viewpoint. Conclusions drawn by participants should be derived from objective analysis of scientific data.
ACCREDITATION STATEMENT
 Pharmacy
Pharmacy
Postgraduate Healthcare Education, LLC is accredited by the Accreditation Council for Pharmacy Education as a provider of continuing pharmacy education.
UAN: 0430-0000-21-112-H01-P
Credits: 2.0 hours (0.20 ceu)
Type of Activity: Knowledge
TARGET AUDIENCE
This accredited activity is targeted to pharmacists. Estimated time to complete this activity is 120 minutes.
Exam processing and other inquiries to:
CE Customer Service: (800) 825-4696 or cecustomerservice@powerpak.com
DISCLAIMER
Participants have an implied responsibility to use the newly acquired information to enhance patient outcomes and their own professional development. The information presented in this activity is not meant to serve as a guideline for patient management. Any procedures, medications, or other courses of diagnosis or treatment discussed or suggested in this activity should not be used by clinicians without evaluation of their patients’ conditions and possible contraindications or dangers in use, review of any applicable manufacturer’s product information, and comparison with recommendations of other authorities.
GOAL
To educate participants about the newly FDA-approved agent aducanumab, including the controversy surrounding its approval.
OBJECTIVES
After completing this activity, the participant should be able to:
- Describe the beta-amyloid theory of Alzheimer’s disease.
- Identify general drug information about aducanumab.
- Recognize the medications currently available to manage Alzheimer’s disease.
- Examine the pharmacist’s role in educating patients about aducanumab.
ABSTRACT: Aducanumab is a monoclonal antibody directed against the beta-amyloid plaques thought to be involved in the development of Alzheimer’s disease. This agent was granted accelerated approval by the FDA despite an almost unanimous vote against approval by the FDA’s expert independent advisory committee; the committee’s opinion was based on uncertainty regarding the strength of evidence of efficacy in phase III clinical trials. Phase IV studies of aducanumab will be conducted to verify the suspected clinical benefit, and the data will be used to determine whether traditional drug approval will be granted. There are no contraindications to aducanumab use, but amyloid-related imaging abnormalities can occur. Other medications for treating dementia include the acetylcholinesterase inhibitors donepezil, galantamine, and rivastigmine and the N-methyl-D-aspartate receptor antagonist memantine. It is important for the pharmacist to be up-to-date on the management of dementia in order to help patients and caregivers make treatment decisions and monitor for adverse events associated with treatment.
Aducanumab (Aduhelm) is a monoclonal antibody that is directed against the beta-amyloid plaques that are one of the proposed mechanisms for the development of Alzheimer’s disease.1-3 On June 7, 2021, the FDA approved aducanumab for the treatment of Alzheimer’s disease.4 The agent was approved through the FDA’s Accelerated Approval Pathway. This process allows for earlier approval of drugs that treat serious conditions and fill an unmet medical need based on a surrogate end point, which is a marker that is thought to predict clinical benefit but in itself is not a measure of clinical benefit. In the case of aducanumab, the surrogate end point is beta-amyloid plaque reduction.4,5 Post approval, the manufacturer must conduct phase IV confirmatory studies to verify the suspected clinical benefit; these data will be used to determine whether traditional drug approval will be granted or whether the drug will be withdrawn from the market.5
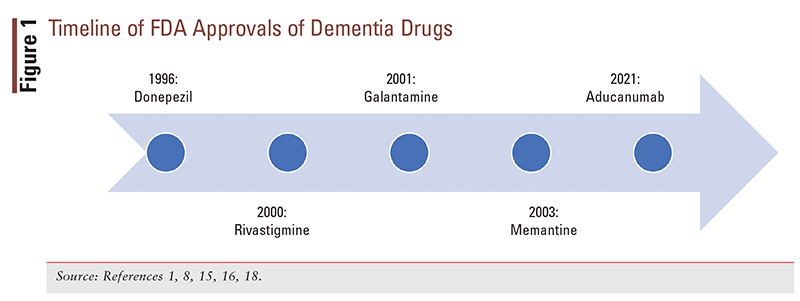
On November 6, 2020, the FDA’s Peripheral and Central Nervous System Advisory Committee voted on whether strong evidence existed to support the effectiveness of aducanumab for the treatment of Alzheimer’s disease. The results of this vote were as follows: yes, 1 vote; no, 8 votes; and uncertain, 2 votes.6 In a letter to the committee chair on June 7, 2021, the FDA explained the rationale for its approval of the drug through the Accelerated Approval Pathway, stating that the effect on the surrogate end point (reduction in beta-amyloid) was met and that the agent was likely to provide a clinical benefit.7 These conflicting opinions have created a great deal of controversy surrounding the approval of this drug. Until the approval of aducanumab, memantine was the most recent (2003) drug approved for Alzheimer’s disease (FIGURE 1).8
Alzheimer’s Disease
More than 6 million (11%) Americans aged 65 years or older have Alzheimer’s disease, and this number is expected to rise as the population ages.2 Alzheimer’s disease is the 6th leading cause of death in the United States, with the rate projected to increase based on trends reported by the National Center for Health Statistics.2,9
There are several theories as to the cause of Alzheimer’s disease, but none have been definitively proven. One of these is that the symptoms of dementia are caused by beta-amyloid plaques that form in the brain. The plaques are composed of aggregates of fragments from the beta-amyloid protein, hence their name. Beta-amyloid plaques, which form between the neurons in the brain, are thought to interfere with neuronal communication at the synapses, thereby contributing to neurodegeneration. Interestingly, autopsy reports show an absence of Alzheimer’s disease–related brain changes in 15% to 30% of patients who presented with symptoms of Alzheimer’s disease. It is theorized that the reduction or removal of beta-amyloid plaques could be beneficial for patients who have Alzheimer’s disease. Another important consideration in the development of Alzheimer’s disease is the presence of apolipoprotein E (APOE). Expression of the gene APOE, specifically the e4 allele, is a strong genetic risk factor for the development of Alzheimer’s disease.2,3,10
Aducanumab
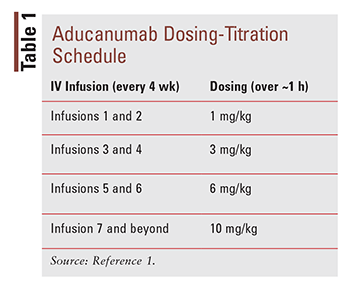
Aducanumab is a human immunoglobulin gamma 1 monoclonal antibody that is expressed in a Chinese hamster ovary cell line.1 Considered a disease-modifying agent, aducanumab is directed against aggregate soluble and insoluble forms of beta-amyloid. It is administered via IV infusion every 4 weeks (the minimum interval is 21 days). A weight-based titration schedule is used, with the maintenance dose being 10 mg/kg (TABLE 1). The IV infusion should be administered over approximately 1 hour. If a dose is missed, it may be administered as soon as possible at the same dosage. The aducanumab solution should have a clear to opalescent quality and look colorless to yellow. Dilution of the solution is required, after which immediate use is recommended. If it cannot be used immediately, the diluted solution may be stored either under refrigeration for up to 3 days or at room temperature for up to 12 hours. The solution is available in two strengths: 170 mg/1.7 mL and 300 mg/3 mL.1
To date, only a small amount of clinical data has been published on the safety and efficacy of aducanumab. Two studies are referenced in the package insert as well as in an editorial by several members of the FDA’s Peripheral and Central Nervous System Advisory Committee on the drug’s effectiveness and information submitted by Biogen to the FDA Advisory Committee: 1) NCT 02484547 (i.e., EMERGE trial); and 2) 02477800 (i.e., ENGAGE trial). These phase III trials were reported to be nearly identical in their methodology (TABLE 2). The EMERGE and ENGAGE trials were terminated halfway through based on results of a futility analysis.1,10,11
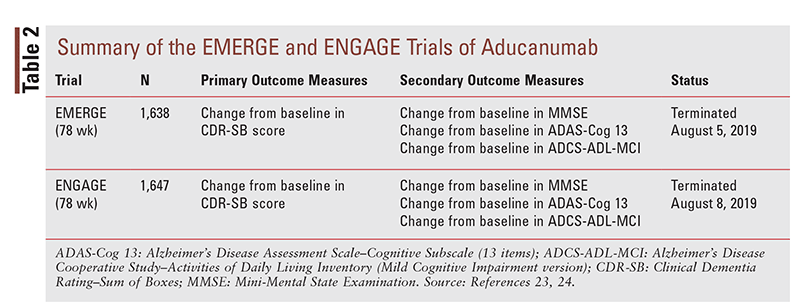
Both of these 78-week randomized, double-blind, placebo-controlled, parallel-group trials, which were conducted in 20 countries worldwide, studied patients aged 50 to 85 years who had mild cognitive impairment or mild dementia with elevated beta-amyloid. An 18-month double-blind, placebo-controlled period was followed by a dose-blinded long-term extension. To be enrolled, patients were required to meet a baseline Mini-Mental State Examination (MMSE) score of 24 to 30 and a Clinical Dementia Rating (CDR) global score of 0.5. Patients who were carriers of APOE e4 and those who were noncarriers were included.10,12
The primary outcome measure for the EMERGE and ENGAGE trials (TABLE 2) was a difference in the CDR Scale–Sum of Boxes (CDR-SB). This scale, which assesses both cognitive effects and daily function, has been shown to have the capability to detect change over time in patients with early symptomatic Alzheimer’s disease. Secondary outcomes included the use of the MMSE, Alzheimer’s Disease Assessment Scale–Cognitive Subscale (13 items), and Alzheimer’s Disease Cooperative Study–Activities of Daily Living Inventory (Mild Cognitive Impairment version) to evaluate clinical decline. Regarding efficacy, the Neuropsychiatric Inventory 10 was a tertiary objective evaluating behavior. Other tertiary objectives were included to assess safety, tolerability, and biomarkers (beta-amyloid and tau levels).
Both high-dose and low-dose aducanumab were studied (TABLE 3), and adjustments were made in Protocol Version 4 to increase the dose in the high-dose arm to 10 mg/kg regardless of APOE e4 carrier status. APOE e4 heterozygosity increases the risk of Alzheimer’s disease three- to fourfold, whereas homozygosity increases the risk by eight to 12 times. There is evidence that the adverse event known as amyloid-related imaging abnormalities (ARIA) occurs more frequently in APOE e4 carriers taking aducanumab, especially at higher doses; this is the reason for the dose stratification based on APOE e4 carrier status.10

After the termination of both studies, Biogen examined individual trial data and found that the EMERGE trial was trending positive with a –18% difference on the CDR-SB, favoring high-dose aducanumab. This trend was not seen in the ENGAGE trial, which demonstrated a 15% difference in CDR-SB favoring placebo. A subsequent analysis of the EMERGE trial that included 3 more months of data found a benefit in all of the primary and secondary trial outcomes for the highdose arm, but not the low-dose arm. Similarly, a subsequent analysis of the ENGAGE trial that included 3 additional months of data showed no benefits in either treatment arm compared with placebo. Why, then, did these two similarly designed trials produce differing outcomes? In a post hoc analysis, Biogen suggested that total duration of high-dose aducanumab played a role. Also, there were fewer participants in the high-dose ENGAGE arm. Additionally, the post hoc analysis found that the placebo groups had a differing decline of dementia, with a larger clinical decline occurring in the EMERGE placebo group (rapid progressor theory).10,12
In their commentary, members of the FDA’s Advisory Committee noted, “Although their exclusion resulted in modest improvement in the estimated efficacy of aducanumab, the sponsor was unable to determine criteria that would prospectively identify such patients to inform future trials or clinical practice, the theory of rapid progressors was introduced post hoc, and statistical analyses to account for them did not change the overall null results of study 301 (ENGAGE).”11 They further stated that post hoc data should not be used as a basis for FDA approval; rather, it should be used to generate hypotheses for future clinical trials, as there are statistical adjustments that occur that can skew data.11
In addition to the clinical data obtained, the trials collected biomarker data via positron emission tomography. These data showed a reduction in beta-amyloid in all treatment groups, but there were no reductions in the placebo groups. In a comparison of the high-dose groups, the EMERGE trial demonstrated a greater degree of reduction than the ENGAGE group (–0.272 vs. –0.238, respectively). The low-dose groups demonstrated beta-amyloid reduction, but the rate was one-half or less that seen in the high-dose groups.10,12
There are no contraindications to aducanumab use, but ARIA can occur. ARIA seen with aducanumab use include edema (ARIA-E) and hemosiderin deposition (ARIA-H); the latter is further categorized as superficial siderosis or microhemorrhage. In the EMERGE and ENGAGE trials, ARIA-E and/or ARIA-H were observed in 41% of patients receiving aducanumab versus 10% of those given placebo. The majority of ARIA-E occurred within eight doses of treatment, with 68% of patients achieving resolution by week 12 and 91% achieving resolution by week 20. Twenty-four percent of patients in the treatment arm who had ARIA presented with clinical symptoms, versus 5% of those in the placebo arm. Headache was the most common complaint, followed by confusion and dizziness. MRI of the brain is recommended within 1 year prior to initiation of treatment in order to determine the development of these abnormalities. Repeat MRI is recommended before the 7th infusion and the 12th infusion.1
Careful monitoring in geriatric patients taking aducanumab is important because this drug is being used in the management of a geriatric syndrome. If a hypersensitivity reaction occurs, immediate discontinuation of the infusion is warranted. Patients up to age 85 years were included in clinical studies, and there were no notable differences in adverse events between age groups. No studies have been conducted in patients with renal or hepatic abnormalities; however, neither renal elimination nor hepatic metabolism is expected to occur. Aducanumab is eliminated via degradation into small peptides and amino acids, with a half-life of 24.8 days. Steady state occurred by 16 weeks of repeated dosing.1
Currently Available Medications Approved for Alzheimer’s Disease
Patients with mild-to-moderate Alzheimer’s disease may be initiated on acetylcholinesterase inhibitor (AChEi) monotherapy. Patients with moderate-to-severe Alzheimer’s disease may be initiated on memantine as monotherapy or in combination with an AChEi. For patients who cannot tolerate AChEis, memantine monotherapy is an option. Should the severity of a patient’s Alzheimer’s disease progress, AChEi discontinuation is not recommended based on disease severity alone.13,14
Three AChEis are currently available for use as monotherapy in mild-to-moderate Alzheimer’s disease: donepezil, galantamine, and rivastigmine (TABLE 4). There is no preferred agent in the class. If one AChEi is tried but fails or is not tolerated, it is acceptable to switch to another agent in the class. The main differences among the agents are their formulations and their adverse effects.13,14
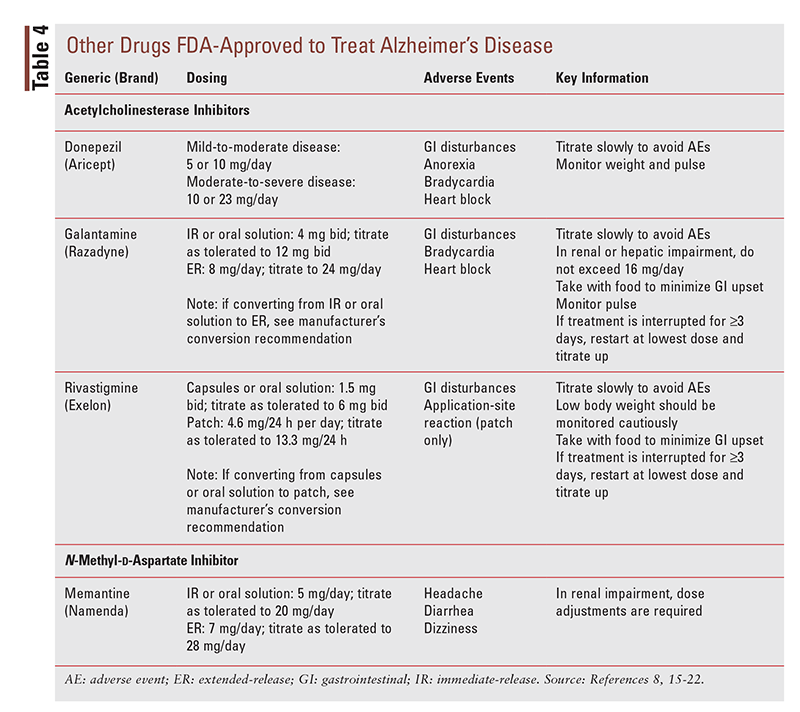
Donepezil: Donepezil (TABLE 4) is available as tablets (5 mg, 10 mg, and 23 mg) and as orally disintegrating tablets (ODTs; 5 mg and 10 mg).15 This agent is FDA approved for use in patients with mild, moderate, or severe Alzheimer’s disease. The dose must be titrated up to avoid side effects. The patient’s dose may be increased from 5 mg to 10 mg after 4 to 6 weeks and from 10 mg to 23 mg after 3 months. The 23-mg dose cannot be split, crushed, or chewed. It is important to note that anorexia may occur, especially at higher doses, so the patient’s weight should be monitored. The patient’s pulse should also be monitored, as bradycardia and heart block have been associated with donepezil use. More commonly, the patient may experience gastrointestinal (GI) disturbances. The ODT formulation should be allowed to completely melt on the tongue. After the tablet has melted, the patient should drink some water.15
Galantamine: Galantamine (TABLE 4) is available as immediate-release tablets (4 mg, 8 mg, and 12 mg), as extended-release tablets (8 mg, 16 mg, and 24 mg), and as an oral solution (4 mg/mL).16,17 It is FDA approved for patients with mild-to-moderate dementia of the Alzheimer’s type. Dosing for the immediate-release tablet and the oral solution starts at 4 mg two times per day, and it may be increased to 8 mg two times per day after 4 weeks. The dosage may be further increased to 12 mg two times per day after 4 weeks on the 8-mg dose if therapy is well tolerated. Dosing for the extended-release tablet starts at 8 mg daily; it may be increased to 16 mg daily after 4 weeks, and again to 24 mg after an additional 4 weeks if it is well tolerated. If therapy is interrupted for 3 or more days, it should be restarted at the lowest dose and retitrated as indicated above. As with donepezil, conduction abnormalities including bradycardia and heart block may occur, and galantamine can contribute to weight loss, so the patient’s pulse and weight should be monitored. The most common side effects of galantamine are GI disturbances, including nausea, vomiting, and diarrhea, and these problems are often the cause of treatment discontinuation. Patients and caregivers should be counseled to administer this agent with food and to ensure adequate hydration during therapy.16,17
Rivastigmine: Rivastigmine (TABLE 4) is available as capsules (1.5 mg, 3 mg, 4.5 mg, and 6 mg), as an oral solution (2 mg/mL), and in patch form (4.6 mg/24 hours, 9.5 mg/24 hours, and 13.3 mg/24 hours).18-20 This agent is FDA approved for the treatment of mild-to-moderate dementia of the Alzheimer’s type; it is also approved for the treatment of Parkinson’s disease dementia, which has a slightly different dosing titration. For the treatment of Alzheimer’s disease, the initial dosage of oral rivastigmine is 1.5 mg two times per day, with titration to 3 mg two times per day after 2 weeks. If treatment is tolerated, the dosage may be increased after another 2 weeks to 4.5 mg two times per day, and finally to 6 mg two times per day after 2 more weeks. Oral rivastigmine should be taken with a meal in order to minimize GI upset.
Treatment with transdermal rivastigmine should be initiated with the 4.6 mg/24 hours patch. The dosage may be titrated after 4 weeks to the 9.5 mg/24 hours patch, and again to the 13.3 mg/24 hours patch after an additional 4 weeks. The patch should be applied to a clean, dry, hairless area that is free of broken skin. If it is likely that the patient will remove the patch, apply it to the upper or lower back; otherwise, the upper arm or the chest may also be used. Patients and caregivers should be advised against cutting the patch, as doing so can affect delivery of the drug. In addition, the adhesive side of the patch should not be touched, as this can cause drug transmission and impair adhesion to the patient’s skin. Application-site reactions may occur with the transdermal formulation of rivastigmine.
The side effects most commonly experienced with rivastigmine use are GI upset (specifically, nausea, vomiting, dyspepsia, diarrhea, and anorexia). The patient’s weight and hydration status should be monitored. Patients with low body weight (<50 kg) are at higher risk for adverse events and should be carefully monitored. If the patient experiences intolerable GI events, rivastigmine should be discontinued for several doses and then restarted at the same dose or a lower dose. If 3 or more days pass between doses, therapy should be restarted at the lowest dose and titrated slowly.18-20
Memantine: Memantine (TABLE 4) is the only N-methyl-d-aspartate receptor antagonist available for the treatment of Alzheimer’s disease; it is currently approved to treat moderate-to-severe disease.8,21,22 Memantine comes in a variety of formulations, including immediate-release tablets (5 mg and 10 mg), extended-release tablets (7 mg, 14 mg, and 21 mg), and oral solution (2 mg/ mL and 10 mg/5 mL). Immediate-release dosing may be initiated at 5 mg once per day and increased by 5 mg every week to a maximum dosage of 20 mg per day. The immediate-release formulation may be given once per day or in two divided doses. The extended-release formulation is initiated at 7 mg per day and increased by 7 mg per week, as tolerated, to a maximum dosage of 28 mg per day. The most commonly reported side effects are headache, diarrhea, and dizziness.8,21,22
The Pharmacist’s Role
Given that they are the most accessible healthcare professionals, pharmacists must be well versed in the evidence presented regarding aducanumab as well as the controversy surrounding the FDA’s approval of this drug. In general, it is important to be up-to-date on the management of dementia in order to help patients and their caregivers make treatment decisions and monitor for adverse events associated with their treatment. Alzheimer’s disease is a difficult disease process for patients and their caregivers. Handling their questions and concerns with knowledge and compassion can help lessen the burden.
REFERENCES
- Aduhelm (aducanumab) package insert. Cambridge, MA: Biogen Inc; July 2021.
- 2021 Alzheimer’s disease facts and figures. Alzheimers Dement. 2021;17(3):327-406.
- Plum F. The pathophysiology of dementia. Gerontology. 1986;32(suppl1):67-72.
- FDA. FDA grants accelerated approval of Alzheimer’s drug. www.fda.gov/ news-events/press-announcements/fda-grants-accelerated-approval-alzheimersdrug. Accessed September 15, 2021.
- FDA. Accelerated approval program. www.fda.gov/drugs/informationhealth-care-professionals-drugs/accelerated-approval-program. Accessed September 15, 2021.
- FDA. Final summary minutes of the Peripheral and Central Nervous System Drugs Advisory Committee meeting. www.fda.gov/media/145690/download. Accessed July 28, 2021.
- Dunn B. Accelerated approval of aducanumab NDA for Alzheimer’s disease [memorandum]. https://fda.report/media/149903/Memorandum+to+PCNS+Chairperson+Nathan+Fountain+regarding+FDA+action+on+aducanu mab.pdf. Accessed September 15, 2021.
- Namenda (memantine) package insert. Madison, NJ: Allergan USA, Inc; November 2018.
- Kochanek KD, Xu J, Arias E. Mortality in the United States, 2019. NCHS Data Brief. 2020;395:1-8.
- Combined FDA and Biogen briefing information for the November 6, 2020, meeting of the Peripheral and Central Nervous System Drugs Advisory Committee. www.fda.gov/media/143502/download. Accessed August 1, 2021.
- Alexander GC, Emerson S, Kesselheim AS. Evaluation of aducanumab for Alzheimer disease: scientific evidence and regulatory review involving efficacy, safety, and futility. JAMA. 2021;325(17):1717-1718.
- Knopman DS, Jones DT, Greicius MD. Failure to demonstrate efficacy of aducanumab: an analysis of the EMERGE and ENGAGE trials as reported by Biogen, December 2019. Alzheimers Dement. 2021;17(4):696701.
- Livingston G, Huntley J, Sommerlad A, et al. Dementia prevention, intervention, and care: 2020 report of the Lancet Commission. Lancet. 2020;396(10248):413-446.
- Dementia: assessment, management and support for people living with dementia and their carers. NICE guideline [NG97]. www.nice.org.uk/guidance/ng97. Accessed September 15, 2021.
- Aricept (donepezil) package insert. New York, NY: Pfizer Inc; February 2012.
- Razadyne (galantamine) package insert. Titusville, NJ: Janssen Pharmaceuticals, Inc; February 2015.
- Galantamine. Lexicomp Online. Waltham, MA: UpToDate, Inc. https:// online.lexi.com. Accessed August 7, 2021.
- Exelon (rivastigmine) package insert. East Hanover, NJ: Novartis Pharmaceuticals Corp; 2016.
- Exelon Patch (rivastigmine transdermal system) package insert. East Hanover, NJ: Novartis Pharmaceuticals Corp; June 2020.
- Rivastigmine. Lexicomp Online. Waltham, MA: UpToDate, Inc. https:// online.lexi.com. Accessed August 7, 2021.
- Memantine. Lexicomp Online. Waltham, MA: UpToDate, Inc. https:// online.lexi.com. Accessed August 7, 2021.
- Namenda XR (memantine) package insert. Madison, NJ: Allergan USA, Inc; November 2019.
- ClinicalTrials.gov. 221AD302 Phase 3 study of aducanumab (BIIB037) in early Alzheimer’s disease (EMERGE). https://clinicaltrials.gov/ct2/show/ record/NCT02484547. Accessed September 15, 2021.
- ClinicalTrials.gov. 221AD301 Phase 3 study of aducanumab (BIIB037) in early Alzheimer’s disease (ENGAGE). https://clinicaltrials.gov/ct2/show/NCT 02477800?term=02477800&draw=2&rank=1. Accessed September 15, 2021.