Antiepileptic Drugs for Epilepsy
RELEASE DATE:
January 1, 2017
EXPIRATION DATE:
January 31, 2019
FACULTY:
Muideen Adigun, PharmD, BCPS, BCPP
Senior Medical Science Liaison
CNS Medical Affairs
Janssen Scientific Affairs, LLC
Washington, DC
Jeremy McLemore, P-4 PharmD Candidate
Howard University College of Pharmacy
Washington, DC
Kasarie Williamson, P-3 PharmD Candidate
Howard University College of Pharmacy
Washington, DC
FACULTY DISCLOSURE STATEMENTS:
The authors declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: Dr. Muideen Adigun is employed by Janssen Scientific Affairs, LLC, a company of Johnson & Johnson. Mr. McLemore and Mr. Williamson have no actual or potential conflicts of interest in relation to this activity.
Postgraduate Healthcare Education, LLC does not view the existence of relationships as an implication of bias or that the value of the material is decreased. The content of the activity was planned to be balanced, objective, and scientifically rigorous. Occasionally, authors may express opinions that represent their own viewpoint. Conclusions drawn by participants should be derived from objective analysis of scientific data.
ACCREDITATION STATEMENT:
Pharmacy
 Postgraduate Healthcare Education, LLC is accredited by the Accreditation Council for Pharmacy Education as a provider of continuing pharmacy education.
Postgraduate Healthcare Education, LLC is accredited by the Accreditation Council for Pharmacy Education as a provider of continuing pharmacy education.
UAN: 0430-0000-17-001-H01-P
Credits: 2.0 hours (0.20 ceu)
Type of Activity: Knowledge
TARGET AUDIENCE:
This accredited activity is targeted to pharmacists. Estimated time to complete this activity is 120 minutes.
Exam processing and other inquiries to:
CE Customer Service: (800) 825-4696 or cecustomerservice@jobson.com
DISCLAIMER:
Participants have an implied responsibility to use the newly acquired information to enhance patient outcomes and their own professional development. The information presented in this activity is not meant to serve as a guideline for patient management. Any procedures, medications, or other courses of diagnosis or treatment discussed or suggested in this activity should not be used by clinicians without evaluation of their patients' conditions and possible contraindications or dangers in use, review of any applicable manufacturer's product information, and comparison with recommendations of other authorities.
GOAL:
To provide pharmacists with an understanding of the antiepileptic drugs (AEDs) used in the treatment of epilepsy and seizure disorders.
OBJECTIVES:
After completing this activity, the participant should be able to:
- Define the difference between a seizure and epilepsy.
- Identify causes and risk factors associated with epilepsy.
- Understand the proposed pathophysiology of seizures.
- Describe the mechanism of action of AEDs used in the management of epilepsy.
- Assess adverse effects and potential drug-drug interactions associated with the use of AEDs.
ABSTRACT: Epilepsy is a neurologic disorder characterized by recurrent, unprovoked seizures that range from shortlived intervals of inattention or muscle jerking to severe and elongated convulsions. The cause of epilepsy is idiopathic in origin in approximately half of diagnosed cases; however, many seizures are either caused naturally or due to brain injuries. Treatment of epilepsy, although difficult, is aimed at eliminating seizure occurrence. Likewise, available pharmacologic agents are associated with a number of adverse effects and drug-drug interactions, making it somewhat challenging when managing patients with epilepsy. When establishing treatment regimens, the aim is to maximize the utilization of antiepileptic drugs (AEDs) while limiting the risk of potential adverse effects or drug interactions.
According to the Epilepsy Foundation, epilepsy is the fourth most common neurologic disorder, affecting approximately 65 million people worldwide.1 In the United States, about 1 in 100 people has had a single unprovoked seizure at some point in their lifetime. Since the advent of antiepileptic drugs (AEDs), about 50% of persons with a new diagnosis of epilepsy become seizure free on their first treatment, with up to 70% becoming seizure free after treatment adjustment.1
Moreover, it is imperative to define the difference between a seizure and epilepsy. A seizure is an isolated event resulting from the clinical manifestation of an abnormal, excessive, hypersynchronous discharge of a population of cortical neurons in the brain, which may produce disturbances in consciousness, motor, sensory, and behavioral activity. Epilepsy, however, is defined as a disorder of the central nervous system (CNS) characterized by recurrent seizures unprovoked by an acute systemic or neurologic insult.2
CLASSIFICATION
The International Classification of Epileptic Seizures classifies seizures as partial (focal), occurring in one hemisphere of the brain, or generalized, occurring in both hemispheres of the brain. Moreover, partial seizures are further broken down to simple partial (patients maintain awareness) and complex partial (impairment of awareness). Generalized seizures are classified into either absence (brief loss of consciousness); myoclonic (sporadic, jerking movements); clonic (repetitive, jerking movements); tonic (muscle stiffness, rigidity); tonic-clonic (formerly grand mal ), or atonic (loss of muscle tone or “drop attacks”) seizures.3
ETIOLOGY
The cause of epilepsy is idiopathic in origin in approximately half of all patients.1 However, several medical conditions have an associated risk or causation with epilepsy, including traumatic brain injury (TBI), CNS infections, hypoglycemia, eclampsia, fever, and stroke. Of note, stroke has been noted as one of the most common medical conditions associated with precipitating seizures. In addition, many medications have been associated with precipitating seizures (TABLE 1).4

PATHOPHYSIOLOGY
It is observed that seizures occur when there is a disturbance in the balance of excitatory and inhibitory neurotransmitters in the CNS. Thus, seizures may occur during hyperactivity, hypofunction, and imbalance of such neurotransmitters. The primary neurotransmitters associated include glutamate (excitatory) and gamma-aminobutyric acid (GABA; inhibitory).2
PHARMACOLOGIC TREATMENT WITH AEDS
With appropriate treatment regimens, typically long- term, a majority of patients with epilepsy live healthy lives and are expected to participate fully in daily activities.2 Thus, the intended goal of treatment is for patients to remain seizure free. This section aims to describe available treatment options, as well as indicate preferred AEDs based on seizure type (TABLE 2).
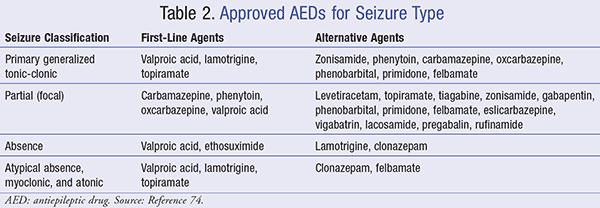
Voltage-Gated Sodium Channel Blockers
Agents within this class play a vital role in the initiation and propagation of action potentials in neurons and excitable cells by mediating the rapid influx of sodium. Various medications, including sodium channel blockers, target inactivated (closed) sodium channels and provide utility in seizure management (TABLE 3).
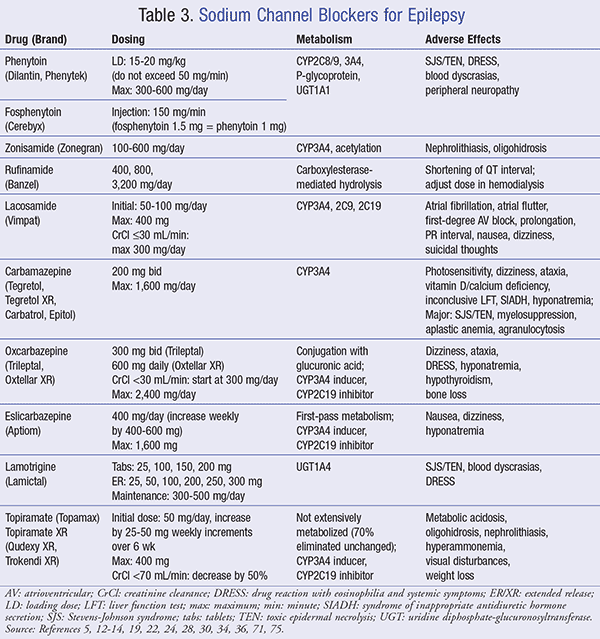
Phenytoin: Phenytoin is among the most widely prescribed AEDs utilized for generalized tonic-clonic seizures, complex partial seizures, mixed seizures, and seizure prophylaxis in neurologic procedures. This agent provides its mechanism by enhancing sodium efflux from neurons of the motor cortex to stabilize the threshold against hyperexcitability.5
A meta-analysis compared phenytoin to carbamazepine for primary outcome of time to withdrawal of allocated treatment and secondary outcomes, including efficacy, recurrence of seizures, and side-effect tolerability.6 The analysis found no clinically significant difference in seizure suppression and seizure recurrence over a 6- to 12-month period between the two agents. However, phenytoin had better side-effect tolerability evidenced by 4% withdrawal of allocated treatment compared to 9% for carbamazepine, a clinically significant result (P = .014).6
Correspondingly, phenytoin is highly protein bound and requires dosage adjustment in patients with hypoalbuminemia and in those who utilize other highly protein-bound medications concurrently.7 Hence, to ascertain the relationship between estimated-free, measured-free, and measured-total phenytoin levels, the Sheiner-Tozer equation is commonly utilized as a predictor for serum phenytoin.7
Prior to initiation, patients should be counseled on the potential for nystagmus, gingival hyperplasia, and rash. Nystagmus occurs in up to 59% of patients who use phenytoin and is an early indicator of toxicity.8 Within 1 to 3 months of initiation, up to 50% of patients can experience phenytoin-induced gingival hyperplasia, with children appearing to be the most susceptible at a prevalence of 88%. Treatment with folic acid at 0.5 mg/day provides an absolute risk reduction of 67%.9-11 Asians and some Caucasians who carry the HLA-B*1502 gene are susceptible to severe exfoliative dermatitis and Stevens-Johnson syndrome (SJS).12
In contrast to the oral formulation, IV phenytoin has been known to precipitate infusion-related adverse cardiovascular effects at rates >50 mg/min. Peripheral catheter administration results in pain, necrosis, and, less commonly, “purple glove syndrome” (i.e., a skin disease in which the extremities become swollen, discolored, and painful).13 Rate reduction to <50 mg/ min and the use of large-gauge catheters is effective in preventing tissue damage attributed to IV phenytoin.
Fosphenytoin: Fosphenytoin, the prodrug of phenytoin, shares similar treatment indications with the additional indication of status epilepticus.14 Of note, status epilepticus is a serious, life-threatening emergency with no universally accepted definition; however, it has been defined as a clinical seizure lasting more than 30 minutes, or repeated seizures over a period of more than 30 minutes without intervening recovery of consciousness.15
Although similar in its efficacy and side-effect profile, fosphenytoin lacks phenytoin’s propylene glycol component, allowing for improved tolerability and decreased incidence of purple glove syndrome associated with IV phenytoin.15 However, in contrast, an identified disadvantage of fosphenytoin is the 10-fold greater acquisition cost compared to phenytoin.16
Zonisamide: Zonisamide, a sulfonamide antiepileptic, is used concomitantly with other anticonvulsants in the treatment of partial seizures. It is structurally unique as it lacks the ureide moiety seen in most sodium channel blockers within its class. In fact, zonisamide was designed with the intention that the sulfamoyl group would provide additional seizures suppression; however, little antiseizure effect has been attributed to this feature at normal doses.17
In a randomized controlled trial of zonisamide as an adjunct agent in partial seizures, evaluating its safety and efficacy, patients treated with zonisamide exhibited a 28.9% reduction in seizure frequency compared to 4.7% in the placebo group over 8 to 12 weeks. Similarly, a study assessing the dose-response of adjunctive zonisamide in 203 patients revealed its efficacy and safety at doses titrated over 5 months (100, 200, and 400 mg/day).18
Rufinamide: Rufinamide is a triazole derivative used as an adjunct to treat Lennox-Gastaut syndrome, a rare form of epilepsy that includes multiple seizure types, in adult and pediatric patients aged >1 year. It modulates sodium channels by prolonging the time spent in the inactivated state, as well as being contraindicated in patients with familial short QT syndrome at doses >2,400 mg.19
Several studies have shown efficacy with rufinamide as add-on treatment in managing drug-resistant epilepsy. A retrospective study conducted in 2014 for children <4 years of age showed a reduction in epileptic spasms and focal seizures by 46% and 30%, respectively.20 Likewise, a meta-analysis of 918 patients was conducted with 521 patients in the rufinamide group and 397 patients in the placebo group. Results reflected significant reduction in the incidence of seizure episodes in 118 patients in the rufinamide group compared to 47 patients in the placebo group.21
Lacosamide: Lacosamide, a functionalized amino acid, possesses dual action against neuronal excitability via slow sodium channel inactivation and collapsin response mediator protein-2 (CRMP2). It is indicated for partial-onset seizures as adjunct and monotherapy in patients aged ≥17 years.22 The agent’s side-effect profile mimics that of most anticonvulsants; however, it provokes unique adverse effects that include atrial fibrillation, atrial flutter, and prolonged PR interval.22 For these reasons, it is recommended that lacosamide be avoided in patients with cardiac conduction abnormalities.
A multicenter open-label trial involving 456 patients was conducted to evaluate the safety and efficacy of lacosamide as first add-on or later adjunct treatment in patients with uncontrolled partial-onset seizures.23 In the first add-on group, 37.5% and 26.5% of patients remained seizure free after 12 and 24 weeks, respectively. In addition, 70.3% experienced >50% reduction in seizure frequency during maintenance therapy. In the later add-on group, 14.9% and 11.6% remained seizure free after completing 12 and 24 weeks of treatment, respectively. Of the 353 patients assessed at the end of the study, 50.4% reported a reduction in seizure frequency with maintenance therapy. Adverse effects were relatively mild with reports of dizziness (33.6%), somnolence (15.0%), and headaches (11.4%).23
Carbamazepine: Carbamazepine, a first-line AED for managing partial and tonic-clonic seizures, has a proposed mechanism of action involving the reduction of polysynaptic responses and blocking post-tetanic potentiation.24 Its first-line recommendation has been supported by comparative research that proves better treatment outcomes compared to other anticonvulsants.25,26 A large, randomized, controlled study conducted by the Veterans Administration group concluded that patients on carbamazepine had a mean composite score that was 9.8 points lower than valproate (P < .005).25 The lower mean composite score reflected lower severity and frequency of partial seizures in the first 12 months of drug therapy.
In relation to its adverse-effect profile, syndrome of inappropriate antidiuretic hormone secretion (SIADH) has been observed in up to 21.7% of patients utilizing high doses of carbamazepine (1,200 mg/day). Additionally, elderly patients are considered high risk for hyponatremia-related SIADH and require dose adjustment or medication cessation.27 Other serious adverse effects, including agranulocytosis and aplastic anemia, although rare, are five times more likely to develop in patients undergoing treatment than the general population.24 Lastly, serious dermatologic complications such as severe rashes, including SJS and toxic epidermal necrolysis (TEN), have been seen with carbamazepine. Prior to treatment, genetic screening to identify high-risk patients, such as those bearing the HLA-B*1502 allele, should be performed. In situations where rash develops, discontinuation should occur immediately.24
Oxcarbazepine: Oxcarbazepine, a derivative of carbamazepine, is indicated for partial and tonic-clonic seizures in adults and children. Although associated with a similar side-effect profile to that of carbamazepine, in some instances oxcarbazepine poses a greater risk for hyponatremia and SIADH as compared to carbamazepine.28 However, oxcarbazepine demonstrates less toxicity and improved efficacy compared to carbamazepine in other areas.28
For example, a systematic review demonstrated a >9% (P = .05) reduction in tonic-clonic seizure frequency with oxcarbazepine when compared to carbamazepine.29 Another trial assessing tolerability and toxicity concluded that oxcarbazepine was superior to carbamazepine with respect to toxicity at high doses.28 The study demonstrated a 50% probability of toxicity at the 1,300 mg carbamazepine dose versus the 2,600 mg dose for oxcarbazepine, thus suggesting more favorable utilization of oxcarbazepine over carbamazepine.
Eslicarbazepine: Eslicarbazepine, a third-generation dibenzazepine, is indicated as monotherapy and adjunctive therapy for partial-onset seizures in adults. It bears structural similarity to both carbamazepine and oxcarbazepine with comparable efficacy and an improved side-effect profile.30 Studies support eslicarbazepine’s efficacy at lower doses, including a controlled phase III trial in 2015 that demonstrated a ≥50% reduction in seizure frequency from baseline after 18 weeks of monotherapy at 1,200 and 1,600 mg.31,32 Indeed, its favorable once-daily dosing increases the potential for improved compliance compared to twice-daily oxcarbazepine and carbamazepine. It should be noted that concurrent use with oxcarbazepine is contraindicated, as eslicarbazepine is the active metabolite of oxcarbazepine, proving a detrimental duplication of therapy.30
The most common adverse events reported by patients taking eslicarbazepine were dizziness (28%) and somnolence (18%). More serious side effects such as ataxia have also been reported in 6% of patients, while hyponatremia and nystagmus were detected in <2% of patients.30 These adverse effects were often attributed to dose increases and ameliorated with reduction. SIADH has been observed in as few as 3% of patients using this medication compared to the higher percentages in oxcarbazepine and carbamazepine.32,33
Lamotrigine: Lamotrigine is indicated as monotherapy or adjunctive therapy for partial seizures, generalized seizures, and Lennox-Gastaut syndrome in adults and children aged ≥2 years. It is believed to inhibit the release of glutamate, an excitatory neurotransmitter via inhibition of voltage-sensitive sodium channels.34 Decreased toxic metabolites, improved CNS adverse effects, and once-daily dosing make lamotrigine a desirable alternative to established therapies.35 Yet, serious skin rashes are its greatest drawback and can elicit life-threatening SJS and TEN. These rashes usually appear within 2 to 8 weeks of starting treatment and are more evident in pediatrics. In the event of rash, lamotrigine should be discontinued and not rechallenged.34
To this end, manufacturers have designed starter kits to decrease the risk for rash. Concurrent use with valproic acid and other inhibitors requires lamotrigine dose reduction by 50%, whereas carbamazepine, phenytoin, and phenobarbital decrease serum level by 40% and demand dose increases. Equally important, protease inhibitors such as lopinavir-ritonavir and atazanavir-ritonavir have been known to decrease lamotrigine concentrations by 50% and 32%, respectively, and may require dose adjustment.34
Topiramate: Topiramate is prescribed as monotherapy and adjunctive therapy for partial-onset seizures, generalized tonic-clonic seizures in children aged >2 years, and Lennox-Gastaut syndrome.36 In partial and generalized new-onset seizures, randomized controlled trials report up to a 76% significant increase (P = .001) in seizure-free rates per 12 months from drug initiation compared to placebo.37,38 Major side effects such as metabolic acidosis occur in up to 67% of patients.36 In addition, oligohidrosis (decreased sweating) is observed, especially in children who account for 10% of reported cases. Lastly, reducing the dose or discontinuing therapy is often enough to prevent long-standing cognitive impairment.39
Drugs That Enhance GABA
GABA is the primary inhibitory neurotransmitter of the CNS, which functions to balance neuronal excitation. An imbalance of this neurotransmitter has been shown to be a probable cause of seizure disorders. To date, a number of medications have proven to be beneficial in the management of seizures by potentiating and preventing the reuptake or metabolism of GABA (TABLE 4).
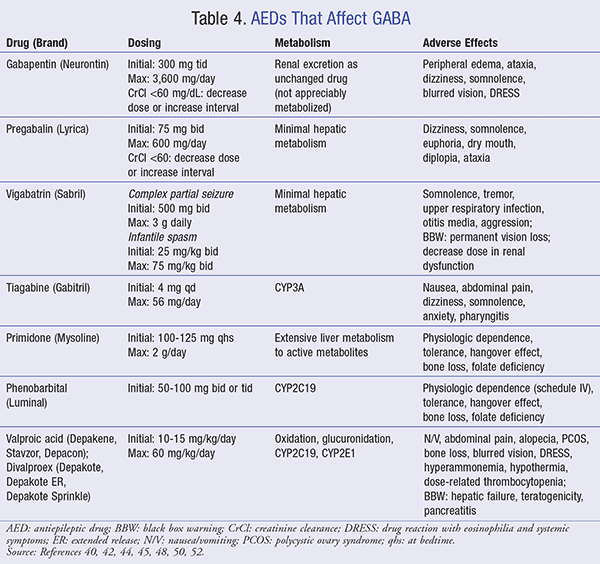
Gabapentin/Pregabalin: Both gabapentin and pregabalin are structurally related to GABA; however, the exact mechanism by which the anticonvulsant effect is exerted is unknown. Gabapentin is used as adjunct therapy for partial seizures in adults and children >3 years of age. One demonstrated advantage of gabapentin over other AEDs is its relatively low toxicities.40 As AED adverse effects are often cognitive in nature, one study shows that gabapentin has no effect on cognition and may offer slight improvement in quality of life based on assessments, including patient survey composite scores, mental health, physical health, and role functioning.41
With regard to pregabalin, it is indicated for adjunct therapy in partial seizures in adults.42 A 2004 study showed that 600 mg daily pregabalin reduced seizure frequency by 42.6% when used as add-on therapy. Investigators also showed that 12% of patients receiving 600 mg daily pregabalin were seizure free during the last month of a 3-month treatment period.43
Vigabatrin: Like other medications in its class, vigabatrin’s exact antiseizure mechanism is unknown. However, this agent is believed to be an irreversible inhibitor of GABA transaminase, the enzyme responsible for GABA metabolism, thus increasing the concentrations of GABA in the CNS. Vigabatrin is indicated as monotherapy for infantile spasm (IS) and as adjunct therapy for refractory (uncontrolled) complex partial seizures in adults. While vigabatrin is a first-line treatment for IS, it is also the only AED considered effective for IS.44
Most importantly, vigabatrin carries a black box warning for permanent vision loss, which increases with increasing dose and cumulative exposure. Visual exams are recommended prior to initiation, every 3 months during, and 3 to 6 months after treatment. For this reason, vigabatrin is only available through the Sabril REMS (Risk Evaluation and Mitigation Strategy) program, formerly known as SHARE, which states that providers and pharmacies must be certified to prescribe and dispense.44
Tiagabine: Tiagabine exerts its antiepileptic effect by inhibiting the presynaptic reuptake of GABA, thus increasing the amount available to bind to postsynaptic receptors. This agent is indicated as adjunct therapy for partial seizures in adults and children 12 years or older.45 Off-label use of tiagabine (i.e., anxiety, neuropathic pain, bipolar disorder) is discouraged due to increased frequency of new-onset seizures or worsening of existing seizures.46
In premarketing trials, the majority of patients were receiving concomitant therapy with at least one enzyme-inducing AED, meaning higher doses of tiagabine had to be given in order to achieve a therapeutic effect.45 In addition, a study published in 2000 showed that patients taking tiagabine for more than 2 years experienced 52% seizure reduction compared to 10.9% reduction in those taking tiagabine for 6 to 12 months, implying a benefit for patients receiving long-term therapy.47
Primidone/Phenobarbital: Barbiturates such as primidone and phenobarbital potentiate GABA-mediated chloride influx by binding to GABAA receptors, allowing chloride (Cl) ions to stabilize and create a less excitable membrane. Primidone is metabolized into phenylethylmalonamide (PEMA) and phenobarbital. While both are active metabolites, PEMA has little antiseizure activity.48
Primidone is indicated for generalized tonic-clonic, psychomotor, and focal seizures.48 Moreover, selected cases have shown that primidone may also be beneficial in patients with QT prolongation and coexisting seizure activity. In support, a literature analysis showed that primidone shortened the QT interval in eight seizure patients with elongated QT intervals and corresponding cardiovascular symptoms.49
Phenobarbital is a schedule IV controlled substance indicated for status epilepticus, generalized tonic-clonic seizures, and partial seizures.50 In the treatment of status epilepticus, IV phenobarbital is indicated as first-line treatment when IV benzodiazepines are not available. It is also important to be aware that extravascular administration of phenobarbital should be avoided due to the risk of necrosis. Additionally, patients with airway obstruction should avoid use due to the increased risk of severe respiratory depression and dyspnea.50,51
Valproic Acid: While its exact mechanism in epilepsy is unknown, valproic acid is thought to act by increasing the amount of GABA in the brain. Equally important, valproic acid is indicated for complex partial seizures and absence seizures (simple and complex).52
A randomized, controlled trial compared valproic acid to lamotrigine and topiramate for generalized and unclassifiable epilepsy with a primary endpoint of time to treatment failure.53 The study concluded that valproic acid was significantly better than topiramate (HR 1.57, 95% CI 1.19-2.08) and indistinguishable from lamotrigine. It was also noted that in patients with idiopathic generalized epilepsy, valproic acid was significantly better than topiramate (HR 1.89, 95% CI 1.32-2.70)and lamotrigine (HR 1.55, 95% CI 1.07-2.24) for the same endpoint.53
In contrast, while valproic acid is widely used, its unfavorable side-effect profile (TABLE 4) is undesirable for many patients. Notably, when used in combination with topiramate, valproic acid can lead to hyperammonemia with or without encephalopathy.54
Benzodiazepines: Benzodiazepines enhance GABA by binding to GABAA receptors, causing a shift in chloride ions, which creates a hyperpolarized (less excitable) neuronal state.55 Currently, six benzodiazepines are utilized in the treatment of epilepsy. These agents are clobazam, clonazepam, clorazepate, diazepam, lorazepam, and midazolam (TABLE 5).
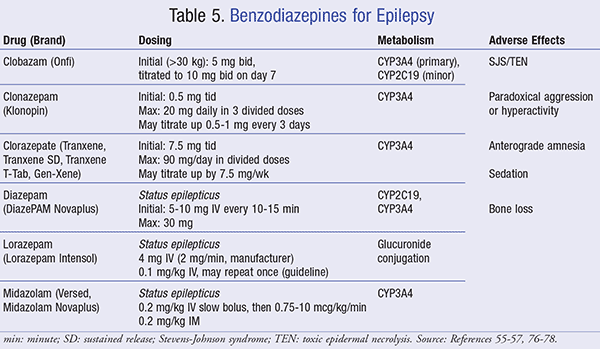
Clobazam is indicated for adjunct therapy in Lennox-Gastaut syndrome.55 Clonazepam is indicated as monotherapy or adjunct treatment for Lennox-Gastaut syndrome, atonic seizures, myoclonic seizures, and absence seizures (in patients who have failed therapy with ethosuximide).56 Clorazepate is indicated for adjunct therapy in partial-onset seizures.57
Diazepam, lorazepam, and midazolam are typically reserved for status epilepticus. To date, lorazepam and diazepam are preferred options in status epilepticus and have no significant differences in effectiveness, while IM midazolam has shown superior effectiveness compared to IV lorazepam in adults with convulsive status epilepticus without established IV access.51
Alternate or Mixed-Mechanism AEDs
Some AEDs have varying mechanisms by which they exerts their effects in the management of epilepsy. These agents include ezogabine, ethosuximide, felbamate, levetiracetam, brivaracetam, and perampanel (TABLE 6).
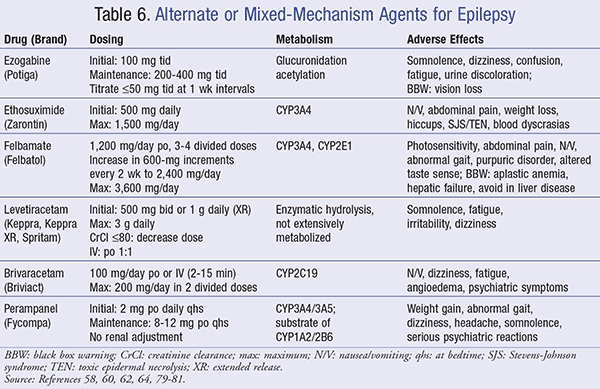
Ezogabine: Approved in 2011, ezogabine is a potassium channel enhancer, which stabilizes neuronal membranes and reduces brain excitability. Ezogabine is indicated for adjunct therapy in partial-onset seizures only in adults with inadequate response to several alternatives.58 In a clinical trial, ezogabine demonstrated a 44.3% reduction in seizure frequency among adults who were refractory to multiple treatments.59
Ezogabine has a black box warning for retinal abnormalities leading to vision loss. Patients receiving ezogabine should have baseline and periodic (every 6 months) visual assessments by an ophthalmic professional. If visual deficits emerge, treatment should be discontinued unless no other suitable choice is available or benefits of use outweigh risks.58
Ethosuximide: Ethosuximide acts by increasing seizure threshold and suppressing paroxysmal spike-and-wave patterns in absence seizures through T-type calcium channel blockade.60 Similar to valproic acid, it is an agent of choice for absence seizures. In a clinical trial, ethosuximide showed no significant difference in efficacy from valproic acid when using freedom from treatment failure after 16 weeks of therapy as the primary endpoint.61
Felbamate: Felbamate is indicated for refractory seizure through the enhancement of GABA and N-methyl-d-aspartate (NMDA) receptor blockers.62 It has demonstrated usefulness as an adjunct in adults and children for the management of partial-onset seizures, Lennox-Gastaut syndrome, and tonic seizure activity. Studies, including a randomized clinical trial, demonstrated a statistically significant reduction in partial-onset seizure in combination with carbamazepine.63 Additionally, there was a 34% reduction in seizure frequency with felbamate as compared to a 9% reduction in the placebo arm.63 Correspondingly, when administered alone, felbamate causes less CNS side effects than other anticonvulsants. However, when combined with other AEDs, it is associated with dose-limiting adverse effects of insomnia, cognitive impairment, and dizziness.63
Levetiracetam: Levetiracetam is indicated for adjunct therapy in myoclonic, partial-onset, and generalized tonic-clonic seizures. Although the mechanism is unknown, it is thought to be multifunctional. It is said that this drug binds to synaptic vesicle protein 2A (SV2A) in the CNS, inhibiting N-type calcium channels, and opposing activity for negative modulators of GABA.64 In a 2007 study, levetiracetam demonstrated its effectiveness as monotherapy for newly diagnosed epilepsy with less withdrawal from adverse events as compared to carbamazepine.65
Different from many AEDs, levetiracetam is classified as Pregnancy Category C. In a prospective cohort study, it was revealed that children exposed to levetiracetam in utero are not at an increased risk of impaired cognitive development within 24 months. Likewise, exposed children were also superior with regard to language and motor development when compared to those exposed to valproic acid, suggesting that levetiracetam may be preferred in women with epilepsy before and during childbearing age.66,67
Brivaracetam: The newest AED to hit the market, brivaracetam is indicated for adjunct therapy in partial-onset seizures in adults aged >16 years.68 Similar to its predecessors, the exact mechanism of anticonvulsant activity is unknown. However, brivaracetam is thought to reduce seizure frequency by binding to SV2A in the brain similar to that of levetiracetam.68
In a randomized, controlled trial utilizing a 28-day seizure frequency as the primary endpoint, brivaracetam therapy resulted in 22.8% and 23.2% reductions in seizure frequency for patients receiving 100 mg/day and 200 mg/day, respectively. Additionally, manufacturers note that no added therapeutic benefit was seen when brivaracetam was coadministered with levetiracetam due to similarities in mechanism of action.68
Perampanel: Utilized in the management of partial-onset seizures, perampanel uses a novel mechanism of action that involves the reduction of neuronal excitation via the noncompetitive antagonism of AMPA (alpha-amino-3-hydroxy-5-methyl-4-isoxazole-propionate) glutamate receptors on postganglionic neurons. Its efficacy has been determined by various trials, including three 19-week, randomized, double-blinded trials, where partial-onset seizure frequency was reduced by up to 35% on maximum dose therapy compared to placebo at 21%.69-71
ROLE OF THE PHARMACIST
Pharmacists play a substantial role in managing patients with epilepsy. More importantly, pharmacists have the ability to assist in educating patients and caregivers about the adverse effects associated with AEDs. In addition to individual side effects, AEDs as a group share many adverse events that include increased risk of suicidality, bone loss (osteoporosis), and severe skin reactions.72,73 Of note, the increased risk of suicidal thoughts/behaviors is a very important classwide risk that pharmacists should be aware of prior to dispensing an AED.73 Equally important, this classwide risk has led to the dispensing of a required Medication Guide in efforts to further counsel patients and caregivers regarding this risk.
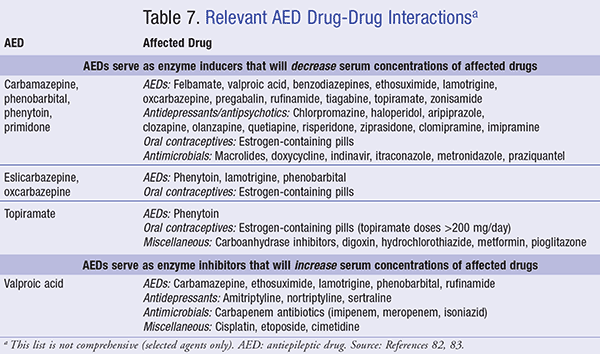
Pharmacists also play a vital role in managing drug- drug interactions with AEDs and medications whose metabolism relies on the CYP450 system. As noted in TABLES 3 to 6, many of the commonly prescribed anticonvulsants are known CYP450 enzyme inducers, including carbamazepine, eslicarbazepine, oxcarbazepine, fosphenytoin, perampanel, phenytoin, phenobarbital, primidone, and topiramate (≥200 mg/day). Accordingly, educating patients and healthcare professionals regarding common drug interactions with AEDs (TABLE 7) and adverse events is key to successfully managing patients with epilepsy.
CONCLUSION
Epilepsy is a complex neurologic condition that requires the use of AEDs to manage symptoms and control seizures. Although many available treatment options do not have exact mechanisms in which seizure control is achieved, current and new treatment modalities have been successful in alleviating seizures in many patients. Ongoing research in the field of epilepsy should prove beneficial, with the goal of allowing patients to reach and maintain longer seizure-free periods.
REFERENCES
- Hirtz D, Thurman DJ, Gwinn-Hardy K, et al. How common are the “common” neurologic disorders? Neurology. 2007;68(5):326-337.
- Bromfield EB, Cavazos JE, Sirven JI, eds. Chapter 1. Basic mechanisms underlying seizures and epilepsy. In: An Introduction to Epilepsy [Internet]. West Hartford, CT: American Epilepsy Society; 2006. www.ncbi.nlm.nih.gov/books/NBK2510/. Accessed December 19, 2016.
- Fisher RS, van Emde Boas W, Blume W, et al. Epileptic seizures and epilepsy: definitions proposed by the International League Against Epilepsy (ILAE) and the International Bureau for Epilepsy (IBE). Epilepsia. 2005;46(4):470-472.
- Thundiyil J, Kearney T, Olson K. Evolving epidemiology of drug-induced seizures reported to a Poison Control Center System. J Med Toxicol. 2007;3(1):15-19.
- Dilantin Infatabs (phenytoin oral chewable tablets) [package insert]. New York, NY: Parke-Davis; 2011.
- Nolan SJ, Marson AG, Weston J, Tudur Smith C. Carbamazepine versus phenytoin monotherapy for epilepsy: an individual participant data review. Cochrane Database Syst Rev. 2015;(8)CD001911.
- Tobler A, Hösli R, Mühlebach S, Huber A. Free phenytoin assessment in patients: measured versus calculated blood serum levels. Int J Clin Pharm. 2016;38(2):303-309.
- Phenytoin sodium injection [package insert]. New York, NY: Pfizer, Parke-Davis; 2011.
- Bharti V, Bansal C. Drug-induced gingival overgrowth: the nemesis of gingiva unravelled. J Indian Soc Periodontol. 2013;17(2):182-187.
- Suneja B, Chopra S, Thomas AM, Pandian J. A clinical evaluation of gingival overgrowth in children on antiepileptic drug therapy. J Clin Diagn Res. 2016;10(1):ZC32-ZC36.
- Arya R, Gulati S, Kabra M, et al. Folic acid supplementation prevents phenytoin-induced gingival overgrowth in children. Neurology. 2011;76(15):1338- 1343.
- Phenytoin sodium [package insert]. New York, NY: Pfizer, Parke-Davis; 2016.
- Chokshi R, Openshaw J, Mehta NN, Mohler E 3rd. Purple glove syndrome following intravenous phenytoin administration. Vasc Med. 2007;12(1):29-31.
- Cerebyx (fosphenytoin sodium injection) [package insert]. New York, NY: Parke-Davis; 2011.
- Thomson A. Fosphenytoin for the treatment of status epilepticus: an evidence-based assessment of its clinical and economic outcomes. Core Evidence. 2005;1(1):65-75.
- Holliday SM, Benfield P, Plosker GL. Fosphenytoin. Pharmacoeconomic implications of therapy. Pharmacoeconomics. 1998;14(6):685-690.
- Leppik IE. Zonisamide: chemistry, mechanism of action, and pharmacokinetics. Seizure. 2004;13(suppl 1): S5-S10.
- Wilfong AA, Willmore LJ. Zonisamide—a review of experience and use in partial seizures. Neuropsychiatr Dis Treat. 2006;2(3):269-280.
- Banzel (rufinamide) oral film-coated tablets, suspension [package insert]. Woodcliff Lake, NJ: Eisai Inc; 2015.
- Grosso S, Coppola G, Dontin SD, et al. Efficacy and safety of rufinamide in children under four years of age with drug-resistant epilepsies. Eur J Paediatr Neurol. 2014;18(5):641-645.
- Verrotti A, Loiacono G, Ballone E, et al. Efficacy of rufinamide in drug-resistant epilepsy: a meta-analysis. Pediatr Neurol. 2011;44(5):347-349.
- Vimpat (lacosamide) oral film coated tablets, oral solution, intravenous injection [package insert]. Smyrna, GA: UCB Inc; 2012.
- Zadeh WW, Escartin A, Byrnes W, et al. Efficacy and safety of lacosamide as first add-on or later adjunctive treatment for uncontrolled partial-onset seizures: a multicentre open-label trial. Seizure. 2015;31:72-79.
- Tegretol (carbamazepine) [package insert]. East Hanover, NJ: Novartis Pharmaceuticals; 2002.
- Mattson RH, Cramer JA, Collins JF. A comparison of valproate with carbamazepine for the treatment of complex partial seizures and secondarily generalized tonic–clonic seizures in adults. N Engl J Med. 1992;327(11):765-771.
- Marson AG, Williamson PR, Clough H, et al; Epilepsy Monotherapy Trial Group. Carbamazepine versus valproate monotherapy for epilepsy: a meta-analysis. Epilepsia. 2002;43(5):505-513.
- Punyawudho B, Ramsay ER, Brundage RC, et al. Population pharmacokinetics of carbamazepine in elderly patients. Ther Drug Monit. 2012;34(2):176-181.
- Trileptal (oxcarbazepine) [package insert] East Hanover, NJ: Novartis Pharmaceuticals; 2011.
- Kalis MM, Huff NA. Oxcarbazepine, an antiepileptic agent. Clin Ther. 2001;23(5):680-700.
- Aptiom (eslicarbazepine acetate) [package insert]. Marlborough, MA: Sunovion Pharmaceuticals Inc; 2015.
- Jacobson MP, Pazdera L, Bhatia P, et al. Efficacy and safety of conversion to monotherapy with eslicarbazepine acetate in adults with uncontrolled partial-onset seizures: a historical-control phase III study. BMC Neurol. 2015;15:46.
- Rocamora R. A review of the efficacy and safety of eslicarbazepine acetate in the management of partial-onset seizures. Ther Adv Neurol Disord. 2015;8(4):178-186.
- Singh RP, Asconape JJ. A review of eslicarbazepine acetate for the adjunctive treatment of partial-onset epilepsy. J Cent Nerv Syst Dis. 2011;3:179-187.
- Lamictal (lamotrigine) [package insert]. Research Triangle Park, NC: GlaxoSmithKline; 2009.
- Cohen AF, Ashby L, Crowley D, et al. Lamotrigine (BW430C), a potential anticonvulsant. Effects on the central nervous system in comparison with phenytoin and diazepam. Brit J Clin Pharmacol. 1985;20(6):619-629.
- Topamax (topiramate) [package insert]. Titusville, NJ: Janssen Pharmaceuticals, Inc; 2009.
- Faught E. Topiramate in the treatment of partial and generalized epilepsy. Neuropsychiatr Dis Treat. 2007;3(6): 811-821.
- Arroyo S, Dodson WE, Privitera MD, et al. Randomized dose-controlled study of topiramate as first- line therapy in epilepsy. Acta Neurologica Scandinavica. 2005;112(4):214-222.
- Fung EL, Nelson EA. Oligohydrosis: underestimated side effect with topiramate treatment. Indian J Pediatr. 2007;74(7):694.
- Neurontin (gabapentin) oral capsules, tablets, solution [package insert]. New York, NY: Parke-Davis; 2009.
- Mortimore C, Trimble M, Emmers E. Effects of gabapentin on cognition and quality of life in patients with epilepsy. Seizure. 1998;7(5):359-364.
- Lyrica (pregabalin) oral capsules, oral solution [package insert]. New York, NY: Pfizer, Inc; 2012.
- Arroyo S, Anhut H, Kugler AR, et al. Pregabalin add-on treatment: a randomized, double-blind, placebo‐ controlled, dose-response study in adults with partial seizures. Epilepsia. 2004;45(1):20-27.
- Sabril (vigabatrin) oral tablets, oral powder for solution [package insert]. Deerfield, IL: Lundbeck; 2015.
- Gabitril (tiagabine HCl) oral tablets [package insert]. North Wales, PA: Teva Pharmaceuticals USA, Inc; 2015.
- Flowers CM, Racoosin JA, Kortepeter C. Seizure activity and off-label use of tiagabine. N Engl J Med. 2006;354(7):773-774.
- Fakhoury T, Uthman B, Abou-Khalil B. Safety of long-term treatment with tiagabine. Seizure. 2000;9(6): 431-435.
- Primidone oral tablets [package insert]. New Castle, DE: Marlex Pharmaceuticals, Inc; 2015.
- Christidis D, Kalogerakis D, Chan TY, et al. Is primidone the drug of choice for epileptic patients with QT-prolongation? A comprehensive analysis of literature. Seizure. 2006;15(1):64-66.
- Phenobarbital oral tablets [package insert]. Eatontown, NJ: West-Ward Pharmaceuticals, Corp; 2006.
- Glauser T, Shinnar S, Gloss D, et al. Evidence-based guideline: treatment of convulsive status epilepticus in children and adults; report of the Guideline Committee of the American Epilepsy Society. Epilepsy Curr. 2016;16(1): 48-61.
- Depakene (valproic acid) oral capsules, oral solution [package insert]. North Chicago, IL: AbbVie Inc; 2013.
- Marson AG, Al-Kharusi AM, Alwaidh M, et al. The SANAD study of effectiveness of valproate, lamotrigine, or topiramate for generalised and unclassifiable epilepsy: an unblinded randomised controlled trial. Lancet. 2007;369(9566):1016-1026.
- Wadzinski J, Franks R, Roane D, Bayard M. Valproate-associated hyperammonemic encephalopathy. J Am Board Fam Med. 2007;20(5):499-502.
- Onfi (clobazam) oral tablets, suspension [package insert]. Deerfield, IL: Lundbeck; 2012.
- Klonopin (clonazepam) oral tablets [package insert]. Princeton, NJ: Sandoz, Inc. 2013.
- Tranxene T-Tab (clorazepate) [package insert]. Barceloneta, PR: Abbott Pharmaceuticals PR Ltd; 2009.
- Potiga (ezogabine) oral tablets [package insert]. Research Triangle Park, NC: GlaxoSmithKline; 2011.
- French JA, Abou-Khalil BW, Leroy RF, et al. Randomized, double-blind, placebo-controlled trial of ezogabine (retigabine) in partial epilepsy. Neurology. 2011;76(18):1555-1563.
- Zarontin (ethosuximide) oral capsules [package insert]. New York, NY: Parke-Davis; 2012.
- Glauser TA, Cnaan A, Shinnar S, et al. Ethosuximide, valproic acid, and lamotrigine in childhood absence epilepsy. N Engl J Med. 2010;362(9):790-799.
- Felbatol (felbamate) [package insert]. Somerset, NJ: Meda Pharmaceuticals Inc; 2011.
- French J, Smith M, Faught E, Brown L. Practice advisory: the use of felbamate in the treatment of patients with intractable epilepsy: report of the Quality Standards Subcommittee of the American Academy of Neurology and the American Epilepsy Society. Neurology. 1999;52(8): 1540-1545.
- Keppra (levetiracetam) oral tablets, oral solution [package insert]. Smyrna, GA: UCB, Inc; 2013.
- Brodie MJ, Perucca E, Ryvlin P, et al. Comparison of levetiracetam and controlled-release carbamazepine in newly diagnosed epilepsy. Neurology. 2007;68(6):402-408.
- Shallcross R, Bromley RL, Irwin B, et al. Child development following in utero exposure: levetiracetam vs sodium valproate. Neurology. 2011;76(4):383-389.
- Shallcross R, Bromley RL, Cheyne CP, et al. In utero exposure to levetiracetam vs valproate: development and language at 3 years of age. Neurology. 2014;82(3):213-221.
- Klein P, Schiemann J, Sperling MR, et al. A randomized, double-blind, placebo-controlled, multicenter, parallel-group study to evaluate the efficacy and safety of adjunctive brivaracetam in adult patients with uncontrolled partial-onset seizures. Epilepsia. 2015;56(12):1890-1898.
- French JA, Krauss GL, Biton V, et al. Adjunctive perampanel for refractory partial-onset seizures: randomized phase III study 304. Neurology. 2012;79(6): 589-596.
- French JA, Krauss GL, Steinhoff BJ, et al. Evaluation of adjunctive perampanel in patients with refractory partial-onset seizures: results of randomized global phase III study 305. Epilepsia. 2013;54(1):117-125.
- Krauss GL, Serratosa JM, Villanueva V, et al. Randomized phase III study 306 adjunctive perampanel for refractory partial-onset seizures. Neurology. 2012;78(18):1408-1415.
- Zaccara G, Franciotta D, Perucca E. Idiosyncratic adverse reactions to antiepileptic drugs. Epilepsia. 2007;48(7):1223-1244.
- Arana A, Wentworth CE, Ayuso-Mateos JL, Arellano F. Suicide-related events in patients treated with antiepileptic drugs. N Engl J Med. 2010;363:542-551.
- Goldenberg MM. Overview of drugs used for epilepsy and seizures: etiology, diagnosis, and treatment. P T. 2010;35(7):392-415.
- Zonegran (zonisamide) oral capsules [package insert]. Woodcliff Lake, NJ: Eisai Inc; 2009.
- Diazepam injection [package insert]. Lake Forest, IL: Hospira Inc; 2004.
- Lorazepam IM, IV injection [package insert]. Lake Forest, IL: Akorn, Inc; 2008.
- Midazolam HCl IM, IV injection solution [package insert]. Lake Forest, IL: Hospira, Inc; 2009.
- Spritam (levetiracetam) oral tablets [package insert]. East Windsor, NJ: Aprecia Pharmaceuticals; 2015.
- Fycompa (perampanel) oral tablets [package insert]. Woodcliff Lake, NJ: Eisai Inc; 2012.
- Briviact (brivaracetam) oral tablets, oral solution, intravenous injection solution [package insert]. Smyrna, GA: UCB, Inc; 2016.
- Perucca E. Clinically relevant drug interactions with antiepileptic drugs. Br J Clin Pharmacol. 2005;61(3): 246-255.
- Johannessen S, Landmark C. Antiepileptic drug interactions—principles and clinical implications. Curr Neuropharmacol. 2010;8:254-267.