Epigenetics and Pharmacoepigenetics: Fine-Tuning Precision Medicine
RELEASE DATE
October 1, 2020
EXPIRATION DATE
October 31, 2022
FACULTY
Donna M. Lisi, PharmD, BCPS, BCACP, BCGP, BCPP
Clinical Pharmacist, Long Term/Post-Acute Care
Hackensack Meridian Health
Eatontown, New Jersey
FACULTY DISCLOSURE STATEMENTS
Dr. Lisi has no actual or potential conflicts of interest in relation to this activity.
Postgraduate Healthcare Education, LLC does not view the existence of relationships as an implication of bias or that the value of the material is decreased. The content of the activity was planned to be balanced, objective, and scientifically rigorous. Occasionally, authors may express opinions that represent their own viewpoint. Conclusions drawn by participants should be derived from objective analysis of scientific data.
ACCREDITATION STATEMENT
 Pharmacy
Pharmacy
Postgraduate Healthcare Education, LLC is accredited by the Accreditation Council for Pharmacy Education as a provider of continuing pharmacy education.
UAN: 0430-0000-20-111-H01-P
Credits: 2.0 hours (0.20 ceu)
Type of Activity: Knowledge
TARGET AUDIENCE
This accredited activity is targeted to pharmacists. Estimated time to complete this activity is 120 minutes.
Exam processing and other inquiries to:
CE Customer Service: (800) 825-4696 or cecustomerservice@powerpak.com
DISCLAIMER:
Participants have an implied responsibility to use the newly acquired information to enhance patient outcomes and their own professional development. The information presented in this activity is not meant to serve as a guideline for patient management. Any procedures, medications, or other courses of diagnosis or treatment discussed or suggested in this activity should not be used by clinicians without evaluation of their patients’ conditions and possible contraindications or dangers in use, review of any applicable manufacturer’s product information, and comparison with recommendations of other authorities.
GOAL
To educate the participant about factors that may contribute to differences in genomic expression throughout the life span and about the impact of age on pharmacoepigenetics.
OBJECTIVES
After completing this activity, the participant should be able to:
- Define what the science of epigenetics encompasses.
- Describe the role of epigenetics in disease manifestation throughout the life span.
- Identify factors that can alter epigenetic expression.
- Recognize the impact of age on pharmacoepigenetics and pharmacokinetics..
ABSTRACT: Epigenetics is an emerging scientific field that studies heritable changes caused by the activation and deactivation of genes without any change in the underlying DNA sequence of the organism. Social determinants of health are thought to play a major role in influencing epigenetics. Although the genome is constant, epigenetics can alter disease expression and occurrence throughout life. Pharmacoepigenetics refers to alterations of pharmacogenetics that are due to epigenetic changes. Gene expression can change throughout the life span, thereby affecting drug therapy. This is accompanied by age-related changes in pharmacokinetics and pharmacodynamics from the perinatal period through adulthood. Pharmacists need to be aware of the latest developments in the field of epigenetics and pharmacoepigenetics in order to optimize disease-state management.
Great strides have been made in the field of genomics since 2015, when President Barack Obama launched the Precision Medicine Initiative (PMI). PMI is “a long-term research endeavor, involving the National Institutes of Health (NIH) and multiple other research centers, which aims to understand how a person’s genetics, environment, and lifestyle can help determine the best approach to prevent or treat disease.”1 The advent of precision medicine has been accompanied by the recognition that additional factors are at play besides those directly related to the genome.
Epigenetics and Disease Expression Throughout the Life Span
Whereas the genome (genetic material of an organism) is thought to be relatively stable throughout life, gene expression and genetic regulation of expression fluctuate in response to environmental exposures, including adverse events in early life.2 Epigenetics, an emerging scientific field, is the study of heritable changes caused by the activation and deactivation of genes without any alteration in the organism’s underlying DNA sequence.3 The epigenome is responsible for the functional use and stability of information within the genome, and it acts to link the genotype (an individual’s collection of genes) to the phenotype (an individual’s observable traits). Epigenetic changes are the missing pieces accounting for differences in patterns of aging between two genetically identical persons.
DNA Methylation, Histone Modification, and Noncoding RNA
As a person ages, sporadic changes occur in the epigenome that are caused by both exogenous and endogenous sources. Epigenetic changes are denoted by alterations in DNA methylation, histone modification, and transcription of regulatory noncoding RNAs such as microRNAs (miRNAs). DNA methylation affects cellular control of gene expression; histone modification affects the accessibility and transcriptional activity of chromatin, which is a complex of DNA and protein (primarily histone protein) in the cell nucleus.4-7 With aging, altered transcription patterns and transcriptional drifts occur that result in genomic instability and DNA mutations. The DNA mutation may be pathogenic in nature, thereby resulting in disease expression.5 Histone turnover may play a critical role in neuronal plasticity and cognition.8 Histone acetylation enhances transcription, and histone deacetylation suppresses transcription.7 Epigenetics may also play a part in the formation of memories—including long-term memories—through the regulation of sensory cortical remodeling.9
DNA methylation is a biomarker for biological aging that appears to preserve the memory of endogenous and exogenous stresses during life.10 It may also be involved in regulating the splicing of messenger RNA (mRNA) precursors, modulating the activity of enhancers (regulatory elements that enhance transcription by recruiting transcription factors and coactivator proteins), and maintaining chromosomal stability. As a person ages, an accumulation of DNA methylation occurs.11-13 In DNA methylation, a methyl group is added to the 5-carbon base in DNA. DNA methylation that occurs mainly on cytosines and is followed by guanine residues is called CpG methylation; this is the major type of methylation.14 Areas that contain a high frequency of CpG sites are called CpG islands.7 DNA methylation can also occur at sites other than CpG sequences, but this occurs only in specific cell types (i.e., pluripotent stem cells, oocytes, neurons, and glial cells) and in plants. Non-CpG methylation increases markedly after the fetal period.14 DNA methylation that occurs in the promoter region of the genome—the part of the genome involved in RNA transcription— results in the reduction or suppression of gene expression.15,16
Different roles for and/or regulation of epigenetic processes exist in human fetal and adult liver specimens, as evidenced by patterns of gene expression and DNA methylation. It has been observed that increased changes in DNA methylation from fetal age to adulthood are unrelated to regulation of gene expression.16 Age-related changes in DNA methylation, which are thought to be mediated in part by reactive oxygen species (ROS), are involved in altered gene expression and disease presentation (cancer, Alzheimer’s disease, diabetes, multiple sclerosis, stroke, Parkinson’s disease, and diseases of premature aging, such as Hutchinson-Gilford’s progeria syndrome and Werner’s syndrome).13,17,18 Parkinson’s disease and Huntington’s disease may be associated with histone modifications or changes in DNA methylation.14 Neuropathologies seen in younger patients, such as autism and schizophrenia, are also believed to be related to abnormalities in DNA methylation.17 Mutations in genes involved in DNA methylation or normal neuronal development can cause other epigenetic disorders, such as Rett’s syndrome, fragile X syndrome, RubinsteinTaybi’s syndrome, Coffin-Lowry’s syndrome, and alpha-thalassemia mental retardation syndrome.14,18 ROS may also mediate age-related changes in mitochondria, which are thought to contribute to neurodegeneration, apoptosis, malignancies, and age-related diseases.19 Changes in other omics measures across the life span are being discovered, such as undulating changes in the proteome profile.20 These findings’ significance is under investigation, but they appear to be factors in the development of age-related diseases and cognitive and physical impairments.
Role of Social Determinants of Health in Epigenetics
Social determinants of health (SDH) not only play a role in an individual’s current health status but also may be involved in that person’s future wellbeing. SDH are conditions in the environments in which people are born, live, learn, work, play, worship, and age that affect a wide range of health, functioning, and quality-of-life outcomes and risks.21,22 The five key domains of SDH are 1) economic stability, 2) education, 3) health and healthcare, 4) neighborhood and built environment, and 5) social and community context.23
Evidence is mounting that socioeconomic deprivation in early life can affect later life, and even future generations. A recent longitudinal study in JAMA Network Open concluded that children who grew up in more socioeconomically disadvantaged neighborhoods had epigenetic alterations in their DNA methylation affecting inflammation, exposure to tobacco smoke, and metabolism of toxic air pollutants that became evident when they were young adults.24 This suggests that policy interventions at the neighborhood level may affect health trajectories as people age.24 More research is warranted in this area.25 It also has been found that living in the most disadvantaged neighborhoods has been associated with an odds ratio of 2.18 for developing Alzheimer’s disease compared with living in more affluent environments.26
SDH and Critical Stages of Development: The Developmental Origins of Health and Disease hypothesis states that early-life exposures during critical stages of development can influence disease outcomes throughout a person’s life span.27,28 Starting in the prenatal period, environmental stimuli such as diet, stress, exercise, chemical exposure (e.g., tobacco smoke, pollution), and exposure to ionizing radiation or ultraviolet light can contribute to this shift, thereby altering an individual’s epigenetics; this may result in transgenerational epigenetic inheritance.5,13,28,29
Poor maternal nutrition (as determined by the quality and quantity of micronutrients and nonnutrient dietary components during pregnancy) and inadequate nutrition during infancy may result in nutritional programming, which is a cumulative process that produces epigenetic changes in gene expression that affect the cell cycle. This may lead to remodeling and altered organ or tissue function, and it may manifest as an increased risk for diseases such as metabolic syndrome and diabetes throughout the life span and into future generations.30
Diet may affect epigenetics by altering the availability of methyl donors used in DNA methylation or by influencing the activity of enzymes involved in DNA methylation. Nutrients that serve as methyl donors or micronutrients that serve as cofactors for enzymatic activity associated with DNA methylation may play a role in epigenetic changes. Such dietary compounds include folic acid, choline, isoflavones, vitamin B2, vitamin B6, vitamin B12, and methionine. Other substances, such as dietary polyphenols and copper, may also affect epigenetics via histone modification.31,32 Effects of DNA methylation may be tissue-specific, so sampling levels in one biological substance may not reflect changes in other areas of the body.33 Learning and memory may be especially vulnerable to the negative effects of poor nutrition in utero, as the brain is the organ that may be most sensitive to maternal nutritional influences. It is postulated that the detrimental effects of undernutrition or overnutrition, coupled with systemic inflammation and hormonal imbalances in the perinatal period, may be irreversible and may contribute to the development of neurologic and psychiatric diseases (e.g., autism, schizophrenia, or Alzheimer’s disease) in later life.34
Additional environmental factors that have been studied in the etiology of autism include advanced parental age, valproate administration, exposure to toxic chemicals (e.g., air pollution, heavy metals, pesticides), presence of maternal diabetes, enhanced glucocorticoid and immune activity, alterations in zinc-copper balance, and the use of selective serotonin reuptake inhibitors (SSRIs). In particular, polychlorinated biphenyls and bisphenols have been associated with alterations in DNA methylation, leading to epigenetic changes that may directly impact the risk of developing autism spectrum disorder.28,35 In addition to maternally related epigenetic effects, evidence is mounting that sperm may also induce transgenerational epigenetic changes.28
SDH and the Role of Stress: The neurogenetics of stress has been the focus of much study.36 Although its role is complex and somewhat controversial, stress throughout the life span, starting with the prenatal period and continuing through adulthood, may induce telomere erosion, which has been tied to mental-health issues such as depression, anxiety, bipolar disorder, posttraumatic stress disorder, and schizophrenia, as well as to accelerated aging. Telomeres are nucleoprotein complexes at the end of eukaryotic chromosomes that are involved in chromosomal protection, integration, and replication as well as longevity. Shortened telomere length is associated with advancing age and increased morbidity and mortality. As telomeres shorten, they reach a critical threshold at which point cell death or senescence ensues.37-40 Both acute and chronic stress may induce neuroepigenetic changes.36
Excessive life stress can also alter the epigenetic landscape by adversely affecting DNA methylation and inducing glucocorticoid signaling via the hypothalamic-pituitary axis, leading to accelerated epigenetic aging, which may further contribute to the development of age-related diseases.33 The induced changes may be passed on to future generations.36,41 Adverse early-childhood experiences may result in increased immune dysregulation into adulthood.42 Newborn telomere length appears to be related to both maternal cortisol production and the infant’s sex.43 Mitochondria may also be involved in these glucocorticoidrelated epigenetic changes.44 On the other hand, preliminary data indicate that conscientiousness in early life—which has been considered a tool for coping with stress—appears to be associated with longer leukocyte telomere length in adulthood.45,46
Smoking and educational attainment have been studied in relation to telomere length.38,47 One study concluded that smoking-associated methylation changes could result in accelerated telomere shortening.38 In a study examining educational attainment and late-life telomere length in black and white adults, benefits from higher attainment, including protection against telomere shortening, were greater in black subjects.47 It has also been demonstrated that inadequate formal education is an independent predictor of accelerated epigenetic (also known as biological) aging and may contribute to poorer health outcomes in persons who are socioeconomically disadvantaged.10 Additionally, lower socioeconomic status has been associated with negative changes in markers of inflammation, including C-reactive protein and fibrinogen, throughout the life span, finally plateauing in older age (75 years and older).48
Other neuroepigenetic changes throughout the life span result from various forms of stress, including prenatal stress, postnatal stress, adult acute stress, fear learning, and adult chronic stress. These changes can alter DNA methylation, histones, and noncoding RNA. The net effect of these alterations includes changes in how individuals respond to stress and anxiety and changes in brain plasticity (brain’s ability to change and adapt as a result of experience).49 Alterations in noncoding RNA are thought to play a role in the pathophysiology of cognitive disorders, as these substances are involved in neural development, maintenance of neuroplasticity, and stress responses.50 Exposure to violence in childhood has been associated with accelerated epigenetic aging, as determined by changes in DNA methylation as well as heart rate under stressful situations, indicating that early negative life exposures can have lasting effects.51 Given concerns over school gun violence, physical isolation due to the COVID-19 pandemic, cyberbullying, social unrest, and other stressors, this is an area that warrants further study.
SDH and the Role of Obesity and Genomic Parameters: Obesity has been associated with accelerated epigenetic aging.33 Maternal obesity may adversely affect cognitive performance and may influence the future development of psychiatric disorders, including affective disorders, attentiondeficit/hyperactivity disorder (ADHD), and autism spectrum disorders. This may be related to alterations in DNA methylation, as levels of folic acid— a cofactor needed for the methyl donor methionine—have been found to be reduced in the amniotic fluid of obese pregnant women.52 It is also speculated that maternal obesity may impact the intrauterine environment, resulting in epigenetic modifications that increase the inflammatory burden; alter developmental programming of adipose tissue; and contribute to chronic disease throughout adulthood.53
Other genomic parameters, such as the gut microbiome, also change throughout the life span and influence disease occurrence and presentation. For example, the acquisition of antibiotic-resistance genes (the resistome) occurs in puberty.54 The maternal microbiome may influence both prenatal development and postnatal development by modulating the child’s immune system and neural circuits and by influencing nutrients used in brain metabolism.55
Pharmacoepigenetics
In addition to the epigenetic changes mentioned earlier, nucleosome remodeling as well as alterations in DNA methyltransferases, histone deacetylases, histone acetylases, histone methyltransferases, and nucleosomal remodeling factors can affect CYP450 enzymes, leading to altered drug metabolism. Pharmacoepigenetics refers to alterations in pharmacogenetics caused by epigenetic changes. It manifests as alterations in gene expression of drug-metabolizing enzymes and transporters that result in interindividual variations in drug response. CYP enzymes contain critical CpG islands. DNA methylation results in downregulation of CYP enzyme mRNA expression.7 CYP genes are also directly regulated by miRNA or indirectly by nuclear receptors through binding of miRNAs (e.g., direct transcriptional regulation of CYP3A4 promoter by peroxisome proliferatoractivated receptor alpha gene).7,56 Methylation increases from the fetal to the postnatal period, whereas miRNA significantly decreases with age in human cortical tissue.18,57 Nuclear receptors are transcription factors activated by steroidal hormones and other lipid compounds.58 The expression of pharmacokinetic phenotypes is regulated by miRNA.7 Epigenetic processes can influence expression of CYP1A1, CYP1B1, CYP2A13, CYP2E1, CYP1W1, CYP24A1, and CYP3A4 as well as uridine 5ʹdiphospho-glucuronosyl transferase (UGT), glutathione-S-transferase (GST), and the adenosine triphosphate–binding cassette (ABC) transporter and solute carrier (SLC) transporter families of drug transporters and nuclear receptors.7,59 Severe psychosocial deprivation in early childhood increases DNA methylation and induces long-term alterations in gene expression and function of CYP2E1, an enzyme involved in the metabolism of substances of abuse (i.e., nicotine and alcohol).60,61
Drugs for which pharmacoepigenetic studies are available include clopidogrel, hypoglycemic agents, sulfonylureas, antidepressants, escitalopram, inhaled corticosteroids, biological agents for psoriasis, fenofibrate, anti–programmed death cell 1, statins, chemotherapy, alkylating agents, imatinib, doxorubicin, gemcitabine, antipsychotics, haloperidol, opioids, growth hormone, and highly active antiretroviral therapy.62-90 Valproate is a pan-inhibitor of histone deacetylase, which alters the expression of genes involved in transcription regulation, cell survival, ion homeostasis, cytoskeletal modifications, and signal transduction, and it may ultimately influence neuroconnectivity.91 With in utero exposure, valproate may contribute to learning disabilities, developmental delays, and possibly autism.35,92 Although it is somewhat controversial, the use of SSRIs during pregnancy may be associated with a small increased risk of developing autism.35 Adverse effects have occurred in infants with the SLC6A4 genotype of the serotonin transporter.29
Pharmacogenomics and Pharmacoepigenetics in Pediatrics: Pharmacogenomic and pharmacoepigenetic studies are especially needed in pediatrics as more medications become available to treat disorders in this age group. Different stages of ontogeny throughout the life span influence CYP450 activity.93 However, conducting pharmacogenomic testing in children involves many ethical, social, and legal challenges, including the potential for employment or insurance-coverage discrimination.94 Furthermore, many of the current clinical-practice guidelines involving genetic testing in children are fraught with limitations, including variability in scoring and lack of uniformity in grading systems, leading to suboptimal quality as well as a lack of focus on the effects of genetics on drug therapy.95 An additional barrier is pediatricians’ lack of knowledge of pharmacogenetics.96 Nonetheless, pharmacogenomic studies in children have been conducted for acute lymphoblastic leukemia, sickle cell disease, rhinitis/ asthma, juvenile idiopathic arthritis, short stature, ADHD, epilepsy, lysosomal-storage diseases with central nervous system involvement, and kidney transplantation.97
The developmental period from newborn to adolescent is dynamic, resulting in large variability in drug response and enzyme-metabolizing capacity. This is further compounded by variations in gene expression that occur with physiological maturity from early childhood through adulthood.4,98 The ontogeny of CYP450 isoforms has been described. Total CYP450 amount and activity are fluid during the fetal and postnatal periods.93 Hines identified three classes of enzyme expression during development: 1) class I enzymes, which are expressed to the greatest extent during the fetal period and are reduced or absent within the first days of life (e.g., CYP3A7); 2) class II enzymes, which have a constant expression throughout the fetal period and undergo small increases in childhood (e.g., CYP3A, CYP2B6, CYP2C19); and 3) class III enzymes, which are poorly expressed during fetal development but increase markedly after birth (e.g., CYP2C9, CYP3A4, CYP2D6).99,100 Among children who are born prematurely, gestational age can further influence these enzymatic expressions. It has been suggested that drug disposition be studied at the time when developmental change is most likely to occur and when the use of a particular drug is most appropriate.100
At birth, an infant’s total CYP450 content is 30% to 60% that of an adult’s content, depending on the isozyme and the time period studied. CYP1A2 is at its nadir at birth and begins to rise during the first month of life, reaching adult levels by age 1 year. CYP2C expression becomes apparent in the first day of life and reaches 40% of adult levels by the second week of life, at which time it plateaus. Similarly, CYP2D6 follows a similar course but stabilizes and plateaus at age 1 month. Of the CYP3A4 isozymes, CYP3A7 is the most abundant.93 Differences are demonstrated by the expression of CYP3A7, which becomes evident at gestation day 50, but the isozyme’s level drops markedly by adulthood. Conversely, CYP3A4 and CYP3A5 start increasing at postnatal day 7 and reach one-third of adult levels by the first month of life. If a clinician were testing just for total CYP3A levels, they would be found to remain constant despite these shifts in gene expression.4,93,98,101
Differences in gene expression during the first few months of life occur in UGT. UGT is involved not only in medication metabolism, but also in the metabolism of endogenous compounds such as bilirubin. From birth to age 14 weeks, levels of UGT increase from 1% to adult levels, significantly altering the metabolism of drugs affected by this liver enzyme.4,98 Common substrates for UGT include acetaminophen, morphine, imipramine, thyroxine, serotonin, and testosterone.102 Age-related changes in drug metabolism between fetal and pediatric developmental stages appear to involve alterations in miRNAs.103 CYP2D6dependent O-demethylation activity is concordant with genotype by the second week of life, but CYP3A4-dependent N-demethylation progresses throughout the first year of life.4
To further complicate the situation, altered pharmacodynamic responses in pediatric patients have been observed for antidepressants, antiepileptics, opioids, immunosuppressants, angiotensin receptor antagonists, QTc interval–prolonging drugs, and oral anticoagulants.104 These alterations play a role in the development of adverse events.105
However, despite evidence of differences in drug disposition, only one-quarter of clinical trials that were entered in a clinical-trials registry indicated that pharmacokinetic studies would be performed.106 This is grossly inadequate; regardless, clinical trials that perform pharmacokinetic or pharmacoepigenetic analyses but do not consider ontogenetic stages of development or tissue source may be failing to provide critical information.107
The Pediatric Research Equity Act (PREA), which was passed in 2003, was subsequently reauthorized with changes in 2007. Under the PREA, the FDA is authorized to require pediatric assessment of some approved drugs or biological products for indications in children; at a minimum, this typically includes pharmacokinetic and pharmacodynamic data and safety studies.108 The PREA is a mandatory program, whereas the Best Pharmaceuticals for Children Act (BPCA) is a voluntary program that authorizes the FDA to request pediatric drug studies of both approved and unapproved pediatric indications for a given drug product if that product has the potential to be prescribed for an indication other than the one for which the manufacturer is applying for FDA approval.109 Goals common to the PREA and BPCA are to make new pediatric information and drug labeling available and to foster appropriate use of medications in pediatric patients.110 Additionally, the Office of Pediatric Therapeutics, which is mandated by Congress, has developed five distinct programs focusing on advances in scientific activities, ethics, medication safety, international collaboration and communication, and the promotion of safe and effective medication use in neonates.111
Despite these advances, a study examining pharmacogenomic biomarker information in FDA-approved pediatric drug labels found that this information may not reflect differences in gene expression and physiological maturation in children because the information is often extrapolated from adult studies. A study analyzed pediatric pharmacogenomic information from FDA-approved pediatric drug labels and information available from PharmGKB (the Pharmacogenomics Knowledge Base), BPCA/PREA trials, and ClinicalTrials.gov.98 As of July 2014, 140 drugs were included in the FDA Table on Genomic Biomarkers in Drug Labels; of these, 56 medications were approved for use in pediatric patients. Among these 56 drugs, 41 had labeled biomarkers that were very important pharmacogenes (VIPs) according to PharmGKB, whereas 38 had pharmacogenomic biomarkers of any type based on FDA-approved drug labeling. VIPs are defined as genes that have extensive demonstrated relationships between variants and drug response or metabolism and are expected to have clinical significance as identified by PharmGKB. The CYP enzymes that were most involved in the metabolism of drugs with pharmacogenomic pediatric FDA-approved drug-labeling included (in descending order) CYP2D6, CYP2C19, CYP2C9, and G6PD. Information provided by the BPCA and PREA was limited to pharmacogenomic data on proton pump inhibitors, in particular, pantoprazole. When all of the factors mentioned were considered, pantoprazole was the only drug labeled with pharmacogenomic information obtained specifically from pediatric trials.98
FDA Table of Pharmacogenetic Associations: On February 25, 2020, the FDA published a table of pharmacogenetic associations for which sufficient scientific evidence was available to indicate that subgroups of patients with certain genetic variants or variant-inferred phenotypes are likely to experience alterations in drug metabolism. The table is organized into three sections: 1) pharmacogenetic associations for which data support therapeutic management recommendations (49 drugs); 2) pharmacogenetic associations for which data indicate a potential impact on safety or response (17 drugs); and 3) pharmacogenetic associations for which data demonstrate a potential impact on pharmacokinetic properties only (36 drugs). When individual drugs in each category are considered separately, of the 102 drugs included, only four— codeine, pantoprazole, pimozide, and tramadol— have pediatric labeling information, and all four fall within the supporting therapeutic management recommendations category. These recommendations are as follows112:
• Codeine—CYP2D6 ultrarapid metabolizer status would result in higher systemic active metabolite concentrations and higher adverse-reaction risk (i.e., life-threatening respiratory depression and death). Codeine is contraindicated in patients aged <12 years.
• Pantoprazole—CYP2C19 poor metabolizer status would result in higher systemic concentrations. Dose reductions should be considered in pediatric patients; however, no dosing adjustment is necessary in adult patients who are CYP2C19 poor metabolizers.
• Pimozide—CYP2D6 poor metabolizer status would result in higher systemic concentrations of pimozide. It should be kept in mind that the dosage should not exceed 0.05 mg/kg in pediatric patients or 4 mg/day in adult patients who are poor metabolizers. The dosage should not be increased earlier than 14 days.
• Tramadol—CYP2D6 ultrarapid metabolizer status would result in higher systemic and breast milk metabolite concentrations, which may cause respiratory depression and death. Tramadol is contraindicated in children aged <12 years and in adolescents following tonsillectomy or adenoidectomy. Breastfeeding is not recommended.
In addition to the current paucity of information about pharmacogenomics in pediatric patients, basic knowledge about how drug-drug interactions affect children is also lacking because much of this information has been extrapolated from adult clinical trials. In children, for example, dissolution, intestinal absorption, and first-pass metabolism of drugs may differ from that in adults because of alterations in gastric pH (gastric pH is neutral at birth), gastric volume, transit time, gastrointestinal solubility, luminal enzymes, gastrointestinal flora, intestinal-wall enzymes, and transporters.107,113-115
Conclusion
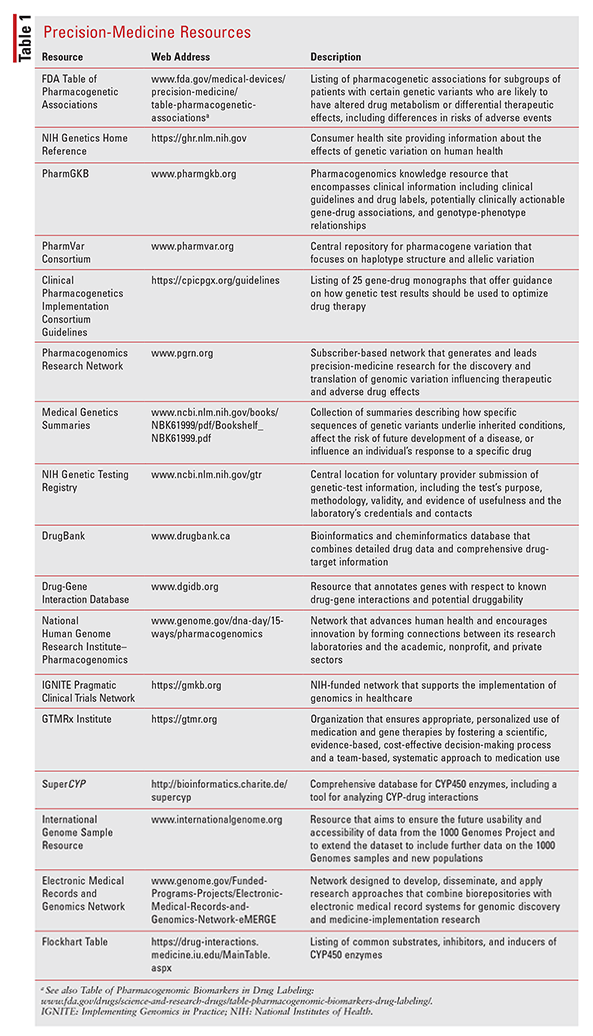
As scientists’ knowledge of epigenetics, pharmacoepigenetics, and age-related pharmacokinetics and pharmacodynamics continues to grow, it is hoped that studies such as the Adult Netherlands Twin Register—which is collecting data on metabolomics, telomere length, and DNA and RNA sequencing—will provide a wealth of new information in the field.116 Information gleaned from epigenetic studies is being used to help calculate polygenic risk scores, which weigh the sum of the number of risk alleles an individual carries to help predict disease course or response to treatment; these scores are beginning to have clinical application (e.g., BRCA1 and BRCA2).117 It is essential for pharmacists to keep abreast of current developments in the fields of epigenetics, pharmacogenetics, and precision medicine. See TABLE 1 for a summary of various resources that can assist in this effort.
REFERENCES
1. Genetics Home Reference. What is the precision medicine initiative? https://ghr.nlm.nih.gov/primer/precisionmedicine/initiative. Accessed June 11, 2020.
2. Balliu B, Durrant M, de Goede O, et al. Genetic regulation of gene expression and splicing during a 10-year period of human aging. Genome Biol. 2019;20(1):230.
3. National Human Genome Research Institute. Epigenetics. www. genome.gov/genetics-glossary/Epigenetics. Accessed June 11, 2020.
4. Zanger UM, Schwab M. Cytochrome P450 enzymes in drug metabolism: regulation of gene expression, enzyme activities, and impact of genetic variation. Pharmacol Ther. 2013;138(1):103-141.
5. Pal S, Tyler JK. Epigenetics and aging. Sci Adv. 2016;2(7):e1600584.
6. ScienceDirect. Chromatin. www.sciencedirect.com/topics/medicine-anddentistry/chromatin. Accessed June 4, 2020.
7. Kim IW, Han N, Burckart GJ, Oh JM. Epigenetic changes in gene expression for drug-metabolizing enzymes and transporters. Pharmacotherapy. 2014;34(2):140-150.
8. Maze I, Wenderski W, Noh KM, et al. Critical role of histone turnover in neuronal transcription and plasticity. Neuron. 2015;87(1):77-94.
9. Phan ML, Bieszczad KM. Sensory cortical plasticity participates in the epigenetic regulation of robust memory formation. Neural Plast. 2016;2016:7254297.
10. Fiorito G, McCrory C, Robinson O, et al. Socioeconomic position, lifestyle habits and biomarkers of epigenetic aging: a multi-cohort analysis. Aging (Albany NY). 2019;11(7):2045-2070.
11. Lev Maor G, Yearim A, Ast G. The alternative role of DNA methylation in splicing regulation. Trends Genet. 2015;31(5):274-280.
12. Angeloni A, Bogdanovic O. Enhancer DNA methylation: implications for gene regulation. Essays Biochem. 2019;63(6):707-715.
13. Rang FJ, Boonstra J. Causes and consequences of age-related changes in DNA methylation: a role for ROS? Biology (Basel). 2014;3(2):403425.
14. Jang HS, Shin WJ, Lee JE, Do JT. CpG and non-CpG methylation in epigenetic gene regulation and brain function. Genes (Basel). 2017;8(6):148.
15. National Human Genome Research Institute. Promoter. www. genome.gov/genetics-glossary/Promoter#:~:text=The%20promoter%20 region%20is%20the,to%20help%20RNA%20get%20transcribed. Accessed June 4, 2020.
16. Huse SM, Gruppuso PA, Boekelheide K, Sanders JA. Patterns of gene expression and DNA methylation in human fetal and adult liver. BMC Genomics. 2015;16:981.
17. Yang J, Yu L, Gaiteri C, et al. Association of DNA methylation in the brain with age in older persons is confounded by common neuropathologies. Int J Biochem Cell Biol. 2015;67:58-64.
18. Muratore CR, Hodgson NW, Trivedi MS, et al. Age-dependent decrease and alternative splicing of methionine synthase mRNA in human cerebral cortex and an accelerated decrease in autism. PLoS One. 2013;8(2):e56927.
19. Gruber J, Fong S, Chen CB, et al. Mitochondria-targeted antioxidants and metabolic modulators as pharmacological interventions to slow ageing. Biotechnol Adv. 2013;31(5):563-592.
20. Lehallier B, Gate D, Schaum N, et al. Undulating changes in human plasma proteome profiles across the lifespan. Nat Med. 2019;25(12):1843-1850.
21. Healthy People 2020. Social determinants of health: overview. www. healthypeople.gov/2020/topics-objectives/topic/social-determinants-ofhealth. Accessed June 4, 2020.
22. CDC. Social determinants of health: know what affects health. www. cdc.gov/socialdeterminants/index.htm. Accessed June 2, 2020.
23. Healthy People 2020. Social determinants of health: interventions & resources. www.healthypeople.gov/2020/topics-objectives/topic/socialdeterminants-health/interventions-resources. Accessed June 2, 2020.
24. Reuben A, Sugden K, Arseneault L, et al. Association of neighborhood disadvantage in childhood with DNA methylation in young adulthood. JAMA Netw Open. 2020;3(6):e206095.
25. Dunn EC. The role of neighborhood social characteristics on the epigenome—why the lack of investigations? JAMA Netw Open. 2020;3(6):e206111.
26. Powell WR, Buckingham WR, Larson JL, et al. Association of neighborhood-level disadvantage with Alzheimer disease neuropathology. JAMA Netw Open. 2020;3(6):e207559.
27. Birnbaum LS, Miller MF. Prenatal programming and toxicity (PPTOX) introduction. Endocrinology. 2015;156(10):3405-3407.
28. Heindel JJ. The developmental basis of disease: update on environmental exposures and animal models. Basic Clin Pharmacol Toxicol. 2019;125(suppl 3):5-13.
29. Blumenfeld YJ, Reynolds-May MF, Altman RB, El-Sayed YY. Maternal-fetal and neonatal pharmacogenomics: a review of current literature.
J Perinatol. 2010;30(9):571-579.
30. Langley-Evans SC. Nutrition in early life and the programming of adult disease: a review. J Hum Nutr Diet. 2015;28(suppl 1):1-14.
31. McKay JA, Mathers JC. Diet induced epigenetic changes and their implications for health. Acta Physiol (Oxf). 2011;202(2):103-118.
32. Bekdash RA. Choline, the brain and neurodegeneration: insights from epigenetics. Front Biosci (Landmark Ed). 2018;23:1113-1143.
33. Gassen NC, Chrousos GP, Binder EB, Zannas AS. Life stress, glucocorticoid signaling, and the aging epigenome: implications for agingrelated diseases. Neurosci Biobehav Rev. 2017;74(Pt B):356-365.
34. Moody L, Chen H, Pan YX. Early-life nutritional programming of cognition—the fundamental role of epigenetic mechanisms in mediating the relation between early-life environment and learning and memory process. Adv Nutr. 2017;8(2):337-350.
35. Bölte S, Girdler S, Marschik PB. The contribution of environmental exposure to the etiology of autism spectrum disorder. Cell Mol Life Sci. 2019;76(7):1275-1297.
36. Griffiths BB, Hunter RG. Neuroepigenetics of stress. Neuroscience. 2014;275:420-435.
37. Shalev I, Entringer S, Wadhwa PD, et al. Stress and telomere biology: a lifespan perspective. Psychoneuroendocrinology. 2013;38(9):18351842.
38. Gao X, Mons U, Zhang Y, et al. DNA methylation changes in response to active smoking exposure are associated with leukocyte telomere length among older adults. Eur J Epidemiol. 2016;31(12):12311241.
39. Savolainen K, Eriksson JG, Kananen L, et al. Associations between early life stress, self-reported traumatic experiences across the lifespan and leukocyte telomere length in elderly adults. Biol Psychol. 2014;97:35-42.
40. Molina-Molina M. Telomere shortening is behind the harm of immunosuppressive therapy in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2019;200(3):274-275.
41. Lebowitz ER, Leckman JF, Silverman WK, Feldman R. Cross-generational influences on childhood anxiety disorders: pathways and mechanisms. J Neural Transm (Vienna). 2016;123(9):1053-1067.
42. Fagundes CP, Glaser R, Kiecolt-Glaser JK. Stressful early life experiences and immune dysregulation across the lifespan. Brain Behav Immun. 2013;27(1):8-12.
43. Bosquet Enlow M, Sideridis G, Bollati V, et al. Maternal cortisol output in pregnancy and newborn telomere length: evidence for sex-specific effects. Psychoneuroendocrinology. 2019;102:225-235.
44. Picard M, McEwen BS, Epel ES, Sandi C. An energetic view of stress: focus on mitochondria. Front Neuroendocrinol. 2018;49:72-85.
45. Edmonds GW, Côté HC, Hampson SE. Childhood conscientiousness and leukocyte telomere length 40 years later in adult women—preliminary findings of a prospective association. PLoS One. 2015;10(7):e0134077.
46. Bartley CE, Roesch SC. Coping with daily stress: the role of conscientiousness. Pers Individ Dif. 2011;50(1):79-83.
47. Adler N, Pantell MS, O’Donovan A, et al. Educational attainment and late life telomere length in the Health, Aging and Body Composition Study. Brain Behav Immun. 2013;27(1):15-21.
48. Davillas A, Benzeval M, Kumari M. Socio-economic inequalities in C-reactive protein and fibrinogen across the adult age span: findings from Understanding Society. Sci Rep. 2017;7(1):2641.
49. Hunter RG, McEwen BS. Stress and anxiety across the lifespan: structural plasticity and epigenetic regulation. Epigenomics. 2013;5(2):177-194.
50. Qureshi IA, Mehler MF. Non-coding RNA networks underlying cognitive disorders across the lifespan. Trends Mol Med. 2011;17(6):337346.
51. Jovanovic T, Vance LA, Cross D, et al. Exposure to violence accelerates epigenetic aging in children. Sci Rep. 2017;7(1):8962.
52. Contu L, Hawkes CA. A review of the impact of maternal obesity on the cognitive function and mental health of the offspring. Int J Mol Sci. 2017;18(5):1093.
53. Tarantal AF, Berglund L. Obesity and lifespan health—importance of the fetal environment. Nutrients. 2014;6(4):1725-1736.
54. Kundu P, Blacher E, Elinav E, Pettersson S. Our gut microbiome: the evolving inner self. Cell. 2017;171(7):1481-1493.
55. Jašarević E, Bale TL. Prenatal and postnatal contributions of the maternal microbiome on offspring programming. Front Neuroendocrinol. 2019;55:100797.
56. Zanger UM, Klein K, Thomas M, et al. Genetics, epigenetics, and regulation of drug-metabolizing cytochrome P450 enzymes. Clin Pharmacol Ther. 2014;95(3):258-261.
57. Beveridge NJ, Santarelli DM, Wang X, et al. Maturation of the human dorsolateral prefrontal cortex coincides with a dynamic shift in microRNA expression. Schizophr Bull. 2014;40(2):399-409.
58. Sever R, Glass CK. Signaling by nuclear receptors. Cold Spring Harb Perspect Biol. 2013;5(3):a016709.
59. Gomez A, Ingelman-Sundberg M. Epigenetic and microRNA-dependent control of cytochrome P450 expression: a gap between DNA and protein. Pharmacogenomics. 2009;10(7):1067-1076.
60. Kumsta R, Marzi SJ, Viana J, et al. Severe psychosocial deprivation in early childhood is associated with increased DNA methylation across a region spanning the transcription start site of CYP2E1. Transl Psychiatry. 2016;6(6):e830.
61. García-Suástegui WA, Ramos-Chávez LA, Rubio-Osornio M, et al. The role of CYP2E1 in the drug metabolism or bioactivation in the brain. Oxid Med Cell Longev. 2017;2017:4680732.
62. Sharma AR, Vohra M, Shukla V, et al. Coding SNPs in hsa-miR1343-3p and hsa-miR-6783-3p target sites of CYP2C19 modulates clopidogrel response in individuals with cardiovascular diseases. Life Sci. 2020;245:117364.
63. Khatami F, Mohajeri-Tehrani MR, Tavangar SM. The importance of precision medicine in type 2 diabetes mellitus (T2DM): from pharmacogenetic and pharmacoepigenetic aspects. Endocr Metab Immune Disord Drug Targets. 2019;19(6):719-731.
64. Karaglani M, Ragia G, Panagopoulou M, et al. Search for pharmacoepigenetic correlations in type 2 diabetes under sulfonylurea treatment. Exp Clin Endocrinol Diabetes. 2019;127(4):226-233.
65. Hack LM, Fries GR, Eyre HA, et al. Moving pharmacoepigenetics tools for depression toward clinical use. J Affect Disord. 2019;249:336346.
66. Lisoway AJ, Zai CC, Tiwari AK, Kennedy JI. DNA methylation and clinical response to antidepressant medication in major depressive disorder: a review and recommendations. Neurosci Lett. 2018;669:14-23.
67. Domschke K, Tidow N, Schwarte K, et al. Pharmacoepigenetics of depression: no major influence of MAO-A DNA methylation on treatment response. J Neural Transm (Vienna). 2015;122(1):99-108.
68. Domschke K, Tidow N, Schwarte K, et al. Serotonin transporter gene hypomethylation predicts impaired antidepressant treatment response. Int J Neuropsychopharmacol. 2014;17(8):1167-1176.
69. Powell TR, Smith RG, Hackinger S, et al. DNA methylation in interleukin-11 predicts clinical response to antidepressants in GENDEP. Transl Psychiatry. 2013;3(9):e300.
70. Wang P, Zhang C, Lv Q, et al. Association of DNA methylation in BDNF with escitalopram treatment response in depressed Chinese Han patients. Eur J Clin Pharmacol. 2018;74(8):1011-1020.
71. Wang AL, Qiu W, DeMeo DL, et al. DNA methylation is associated with improvement in lung function on inhaled corticosteroids in pediatric asthmatics. Pharmacogenet Genomics. 2019;29(3):65-68.
72. Ovejero-Benito MC, Reolid A, Sánchez-Jiménez P, et al. Histone modifications associated with biological drug response in moderate-tosevere psoriasis. Exp Dermatol. 2018;27(12):1361-1371.
73. Wei R, Wu Y. Modification effect of fenofibrate therapy, a longitudinal epigenomic-wide methylation study of triglycerides levels in the GOLDN study. BMC Genet. 2018;19(suppl 1):75.
74. Duruisseaux M, Martínez-Cardús A, Calleja-Cervantes ME, et al. Epigenetic prediction of response to anti-PD-1 treatment in non-smallcell lung cancer: a multicentre, retrospective analysis. Lancet Respir Med. 2018;6(10):771-781.
75. Zambrano T, Salazar LA. microRNAs and response to statins in patients with hypercholesterolemia: from basic research to precision medicine. Pharmacogenomics. 2018;19(9):748ć751.
76. Gutierrez-Camino A, Umerez M, Santos B, et al. Pharmacoepigenetics in childhood acute lymphoblastic leukemia: involvement of miRNA polymorphisms in hepatotoxicity. Epigenomics. 2018;10(4):409417.
77. Umerez M, Garcia-Obregon S, Martin-Guerrero I, et al. Role of miRNAs in treatment response and toxicity of childhood acute lymphoblastic leukemia. Pharmacogenomics. 2018;19(4):361-373.
78. Fornaro L, Vivaldi C, Caparello C, et al. Pharmacoepigenetics in gastrointestinal tumors: MGMT methylation and beyond. Front Biosci (Elite Ed). 2016;8:170-180.
79. Crea F, Di Paolo A, Liu HH, et al. Polycomb genes are associated with response to imatinib in chronic myeloid leukemia. Epigenomics. 2015;7(5):757-765.
80. Flanagan JM, Wilhelm-Benartzi CS, Metcalf M, et al. Association of somatic DNA methylation variability with progression-free survival and toxicity in ovarian cancer patients. Ann Oncol. 2013;24(11):2813-2818.
81. Dejeux E, Rønneberg JA, Solvang H, et al. DNA methylation profiling in doxorubicin treated primary locally advanced breast tumours identifies novel genes associated with survival and treatment response. Mol Cancer. 2010;9:68.
82. Candelaria M, de la Cruz-Hernández E, Pérez-Cárdenas E, et al. Pharmacogenetics and pharmacoepigenetics of gemcitabine. Med Oncol. 2010;27(4):1133-1143.
83. Swathy B, Saradalekshmi KR, Nair IV, et al. Understanding the influence of antipsychotic drugs on global methylation events and its relevance in treatment response. Epigenomics. 2018;10(3):233-247.
84. Swathy B, Banerjee M. Understanding epigenetics of schizophrenia in the backdrop of its antipsychotic drug therapy. Epigenomics. 2017;9(5):721-736.
85. Swathy B, Saradalekshmi KR, Nair IV, et al. Pharmacoepigenomic responses of antipsychotic drugs on pharmacogenes are likely to be modulated by miRNAs. Epigenomics. 2017;9(6):811-821.
86. Swathy B, Banerjee M. Haloperidol induces pharmacoepigenetic response by modulating miRNA expression, global DNA methylation and expression profiles of methylation maintenance genes and genes involved in neurotransmission in neuronal cells. PLoS One. 2017;12(9):e0184209.
87. Chu SK, Yang HC. Interethnic DNA methylation difference and its implications in pharmacoepigenetics. Epigenomics. 2017;9(11):14371454.
88. Knothe C, Oertel BG, Ultsch A, et al. Pharmacoepigenetics of the role of DNA methylation in μ-opioid receptor expression in different human brain regions. Epigenomics. 2016;8(12):1583-1599.
89. Ouni M, Belot MP, Castell AL, et al. The P2 promoter of the IGF1 gene is a major epigenetic locus for GH responsiveness. Pharmacogenomics J. 2016;16(1):102-106.
90. Ay E, Banati F, Mezei M, et al. Epigenetics of HIV infection: promising research areas and implications for therapy. AIDS Rev. 2013;15(3):181-188.
91. Rosenzweig I, Vukadinovic Z, Turner AJ, Catani M. Neuroconnectivity and valproic acid: the myelin hypothesis. Neurosci Biobehav Rev. 2012;36(8):1848-1856.
92. Bech LF, Polcwiartek C, Kragholm K, et al. In utero exposure to antiepileptic drugs is associated with learning disabilities among offspring. J Neurol Neurosurg Psychiatry. 2018;89(12):1324-1331.
93. Fanni D, Ambu R, Gerosa C, et al. Cytochrome P450 genetic polymorphism in neonatal drug metabolism: role and practical consequences towards a new drug culture in neonatology. Int J Immunopathol Pharmacol. 2014;27(1):5-13.
94. Haga SB. Pharmacogenomic testing in pediatrics: navigating the ethical, social, and legal challenges. Pharmgenomics Pers Med. 2019;12:273285.
95. Jiao XF, Li HL, Cheng L, et al. Methodological quality of clinical practice guidelines for genetic testing in children: a systematic assessment using the appraisal of guidelines for research and evaluation II instrument. Medicine (Baltimore). 2019;98(52):e18521.
96. Rahawi S, Naik H, Blake KV, et al. Knowledge and attitudes on pharmacogenetics among pediatricians. J Hum Genet. 2020;65(5):437444.
97. Stevens A, De Leonibus C, Hanson D, et al. Pediatric perspective on pharmacogenomics. Pharmacogenomics. 2013;14(15):1889-1905.
98. Kim T, Han N, Sohn M, et al. Pharmacogenomic biomarker information in FDA-approved paediatric drug labels. Basic Clin Pharmacol Toxicol. 2015;116(5):438-444.
99. Sadler NC, Nandhikonda P, Webb-Robertson BJ, et al. Hepatic cytochrome P450 activity, abundance, and expression throughout human development. Drug Metab Dispos. 2016;44(7):984-991.
100. Job KM, Gamalo M, Ward RM. Pediatric age groups and approach to studies. Ther Innov Regul Sci. 2019;53(5):584-589.
101. Sim SC, Edwards RJ, Boobis AR, et al. CYP3A7 protein expression is high in a fraction of adult human livers and partially associated with the CYP3A7*1C allele. Pharmacogenet Genomics. 2005;15(9):625-631.
102. Liu W, Ramírez J, Gamazon ER, et al. Genetic factors affecting gene transcription and catalytic activity of UDP-glucuronosyltransferases in human liver. Hum Mol Genet. 2014;23(20):5558-5569.
103. Burgess KS, Philips S, Benson EA, et al. Age-related changes in microRNA expression and pharmacogenes in human liver. Clin Pharma col Ther. 2015;98(2):205-215.
104. Mulla H. Understanding developmental pharmacodynamics: importance for drug development and clinical practice. Paediatr Drugs. 2010;12(4):223-233.
105. van den Anker J, Reed MD, Allegaert K, Kearns GL. Developmental changes in pharmacokinetics and pharmacodynamics. J Clin Pharma col. 2018;58(suppl 10):S10-S25.
106. Viergever RF, Rademaker CM, Ghersi D. Pharmacokinetic research in children: an analysis of registered records of clinical trials. BMJ Open. 2011;1(1):e000221.
107. Nicolas JM, Bouzom F, Hugues C, Ungell AL. Oral drug absorption in pediatrics: the intestinal wall, its developmental changes and current tools for predictions. Biopharm Drug Dispos. 2017;38(3):209-230.
108. CDER World. Pediatric Research Equity Act (PREA). www.accessdata.fda.gov/scripts/cderworld/index.cfm?action=newdrugs:main&unit=4 &lesson=1&topic=6). Accessed June 4, 2020.
109. CDER World. Best Pharmaceuticals for Children Act (BPCA). www.accessdata.fda.gov/scripts/cderworld/index.cfm?action=newdrugs:m ain&unit=4&lesson=1&topic=7). Accessed June 4, 2020.
110. CDER World. Pediatric Research Equity Act vs. Best Pharmaceuticals for Children Act (PREA vs. BPCA). www.accessdata.fda.gov/scripts/ cderworld/index.cfm?action=newdrugs:main&unit=4&lesson=1&to pic=8). Accessed June 4, 2020.
111. FDA. Office of Pediatric Therapeutics. www.fda.gov/about-fda/ office-clinical-policy-and-programs/office-pediatric-therapeutics. Accessed June 4, 2020.
112. FDA. Table of pharmacogenetic Associations. www.fda.gov/medical-devices/precision-medicine/table-pharmacogenetic-associations. Accessed June 2, 2020.
113. Salem F, Johnson TN, Barter ZE, et al. Age related changes in fractional elimination pathways for drugs: assessing the impact of variable ontogeny on metabolic drug-drug interactions. J Clin Pharmacol. 2013;53(8):857-865.
114. Maharaj AR, Edginton AN, Fotaki N. Assessment of age-related changes in pediatric gastrointestinal solubility. Pharm Res. 2016;33(1):52-71.
115. Salem F, Rostami-Hodjegan A, Johnson TN. Do children have the same vulnerability to metabolic drug–drug interactions as adults? A critical analysis of the literature. J Clin Pharmacol. 2013;53(5):559-566.
116. Willemsen G, Vink JM, Abdellaoui A, et al. The Adult Netherlands Twin Register: twenty-five years of survey and biological data collection. Twin Res Hum Genet. 2013;16(1):271-281.
117. Lewis CM, Vassos E. Polygenic risk scores: from research tools to clinical instruments. Genome Med. 2020;12(1):44.