Precision Medicine and Alzheimer's, Parkinson's, and Huntington's Diseases: From Pathogenesis to Emerging Therapeutics
RELEASE DATE
January 1, 2020
EXPIRATION DATE
January 31, 2022
FACULTY
Donna M. Lisi, PharmD, BCPS, BCGP, BCACP, BCPP
Clinical Pharmacist, Long-Term Care
Hackensack Meridian Health
Eatontown, New Jersey
FACULTY DISCLOSURE STATEMENTS
Dr. Lisi has no actual or potential conflict of interest in relation to this activity.
Postgraduate Healthcare Education, LLC does not view the existence of relationships as an implication of bias or that the value of the material is decreased. The content of the activity was planned to be balanced, objective, and scientifically rigorous. Occasionally, authors may express opinions that represent their own viewpoint. Conclusions drawn by participants should be derived from objective analysis of scientific data.
ACCREDITATION STATEMENT
Pharmacy
 Postgraduate Healthcare Education, LLC is accredited by the Accreditation Council for Pharmacy Education as a provider of continuing pharmacy education.
Postgraduate Healthcare Education, LLC is accredited by the Accreditation Council for Pharmacy Education as a provider of continuing pharmacy education.
UAN: 0430-0000-20-004-H01-P
Credits: 2.0 hours (0.20 ceu)
Type of Activity: Knowledge
TARGET AUDIENCE
This accredited activity is targeted to pharmacists. Estimated time to complete this activity is 120 minutes.
Exam processing and other inquiries to:
CE Customer Service: (800) 825-4696 or cecustomerservice@powerpak.com
DISCLAIMER:
Participants have an implied responsibility to use the newly acquired information to enhance patient outcomes and their own professional development. The information presented in this activity is not meant to serve as a guideline for patient management. Any procedures, medications, or other courses of diagnosis or treatment discussed or suggested in this activity should not be used by clinicians without evaluation of their patients' conditions and possible contraindications or dangers in use, review of any applicable manufacturer's product information, and comparison with recommendations of other authorities.
GOAL
To educate pharmacists on the impact of genetics on neurodegenerative disease (NDD) and how drugs are being developed to address these underlying pathologies.
OBJECTIVES
After completing this activity, the participant should be able to:
- Describe the mechanisms involved in the pathogenesis of inherited NDDs such as Alzheimer's disease (AD), Parkinson's disease (PD), and Huntington's disease (HD).
- Discuss the role of genetics in early-onset versus lateonset AD; early-onset adult, late-onset adult, and juvenile-onset PD; and HD.
- Identify the role of genetic testing in AD, PD, and HD. 4. List the various medications in development for NDDs and how they target underlying inherited defects.
- List the various medications in development for NDDs and how they target underlying inherited defects.
ABSTRACT: Neurodegenerative disorders (NDDs) are neurological conditions that are associated with the progressive loss of neurons resulting in motor and/or cognitive deterioration, often leading to death. Alzheimer's disease (AD), the most common cause of dementia, is divided into early-onset familial AD, in which the genes, PSEN1, APP, and PSEN2, play a role, and late-onset familial AD, in which APOEε4 may serve as a risk modifier. Numerous genes have been associated with the development of early-onset, late-onset, and juvenile-onset Parkinson's disease. However, with both of these NDDs, environmental and other factors may play a greater role than genetics for most patients. Huntington's disease (HD) is a monogenic NDD. The causal link between the mutant huntingtin protein and the clinical manifestations of HD is well established. For each of these NDDs, precision medicine is helping to develop targeted treatments, which may one day lead to a cure for these debilitating conditions.
Neurodegenerative disorders (NDDs) are neurological conditions that are associated with the progressive loss of neurons resulting in motor and/or cognitive deterioration, often leading to death.1-3
Numerous mechanisms and shared pathogenic cascades are thought to be involved in the development of NDDs, including the misfolding of proteins, aberrant chaperone behavior and autophagy maintenance (which helps rid the body of mutated proteins), mitochondrial dysfunction, and environmental factors.2 A major common feature leading to neurodegeneration that is shared by NDDs is the deposition of abnormal proteins that have altered physiochemical properties in both the brain and the periphery. This results in the loss of neurons and synapses in specific brain regions producing the characteristic features of each disease state.
In Alzheimer's disease (AD), there are abnormalities in beta-amyloid resulting in plaques as well as inclusions of hyperphosphorylated microtubulebinding tau. Parkinson's disease (PD) is characterized by deposits of alpha-synuclein. Huntington's disease (HD) is identified by polyglutamine protein aggregates.
From the study of families with high penetrance of the mutation and consistent clinical presentations (phenotypes), much can be gleaned about the genetic component of these disease states.4 Precision medicine is offering new hope for patients with these debilitating diseases by unraveling the mechanisms of pathogenesis and assisting in the development of targeted therapies. This article focuses on the genetic etiology of AD, PD, and HD, as well as targeted therapies that are currently in development.
ALZHEIMER'S DISEASE
AD, the most common form of dementia, is an NDD characterized by gradual and progressive loss of cognitive and functional abilities that is often accompanied by behavioral disturbances.5 AD is the sixth leading cause of death in the United States. Currently, there are 5.8 million Americans with AD (or about one in 10 older adults), and this is expected to climb to 13.8 million by 2050.6 Of these patients, 15% to 25% have late-onset familial AD (LOFAD) (i.e., onset after age 60-65 years), and less than 2% have early-onset familial AD (EOFAD; i.e., onset before age 60-65 years), and less than 1% have AD associated with Down syndrome; for approximately 75% the cause of AD is unclear, but it may involve a combination of genetic and environmental factors.7
Familial and nonfamilial AD have the same clinical presentation, and the only way to distinguish them is through a thorough history of the patient (examining for age of onset of symptoms) and family (to determine if there are three or more affected relatives). Genetic variations play a role in the neuropathologic presentation of AD, which includes the appearance of beta-amyloid plaques—intraneuronal neurofibrillary tangles made up of tau protein—and amyloid angiopathy. This is especially true for patients younger than age 50 years.7
Much focus has been placed on the APOEε4 allelle and its role in LOFAD. APOEε4 is one of three major allele variants, which include APOEε2, APOEε3, and APOEε4; APOEε2 demonstrates neuroprotective properties. Being heterozygous (APOEε3/APOEε4) or homozygous (APOEε4/APOEε4) for the allele increases the risk for both EOFAD and LOFAD. However, other factors (e.g., environmental, lifestyle) besides genetics may also contribute to the development of AD, since being either heterozygous or homozygous for the APOEε4 allele doesn't cause AD; onequarter to one-fifth of the general population have either of these genotypes and may be asymptomatic. Even among AD patients, only about 20% to 65% of LOFAD patients are positive for these alleles. Heterozygous status increases the risk of developing LOFAD by about threefold and this is increased to 15-fold among those who are homozygous for the APOEε4 allele. This translates to an increased risk of about 10% to 20% of developing LOFAD by age 75 years if a patient is heterozygous for APOEε4, compared with an elevated risk of 25% to 35% for those homozygous for the allele. Overall, first-degree relatives of a person with LOFAD have a cumulative lifetime risk of developing AD of about 15% to 25%, which is approximately 1.5 to 2 times greater than the risk of the general population.7
Additionally, susceptibility genes, which are variants that do not cause AD but are thought to increase the risk of developing the disease, have been identified. TABLE 1 includes information on specific chromosomes and genes that have been associated with the development of AD, PD, and HD. It also has data on risk-factor genes that may contribute to the manifestation of these diseases and lists gene and/or allele-specific comments. The presence of these genes is not assessed when performing genomic testing because they are not a causative factor in AD. However, the presence of TREM2 (triggering receptor expressed on myeloid cells 2) is a significant risk factor for LOFAD, increasing the odds of developing AD threefold.7
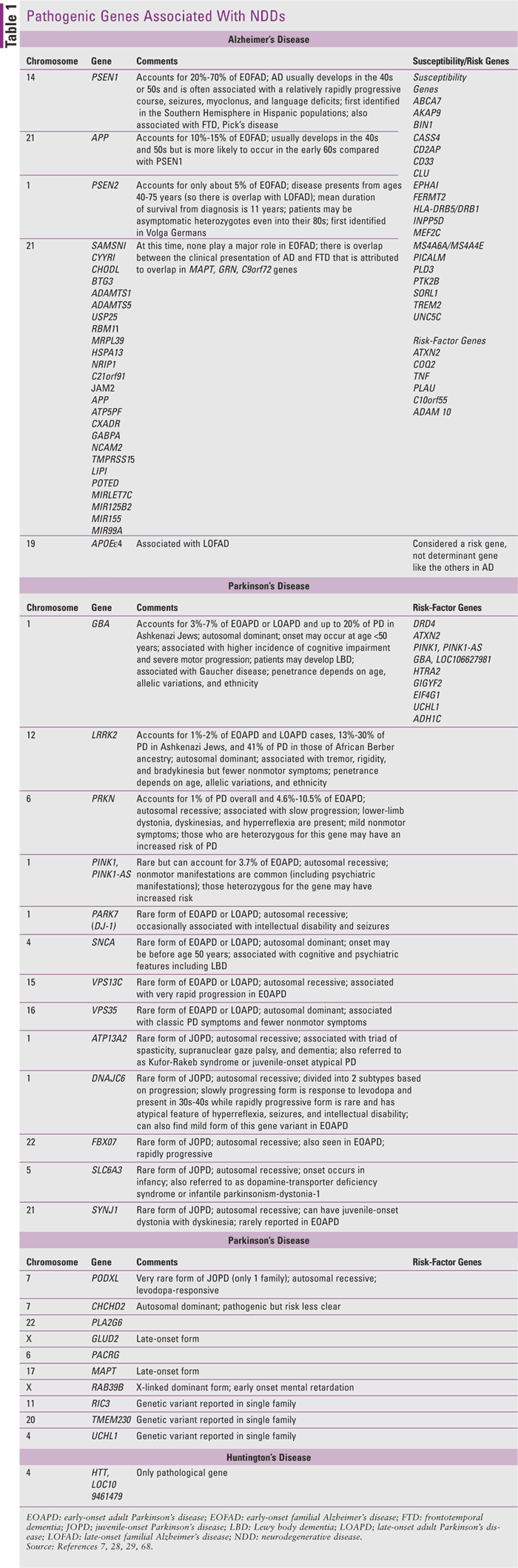
Although the majority of familial AD is due to LOFAD, there is a small group of patients with EOFAD, whose symptoms develop before age 65. About 60% to 80% of EOFAD is accounted for by the presence of three genes, PSEN1 (presenilin 1), APP (amyloid precursor protein) and PSEN2 (presenilin 2)7 (TABLE 1). Alterations in the PSEN1 gene are the most common mutations identified in EOFAD. PSEN1 encodes for presenilin 1, which is a subunit of γ (gamma)-secretase. This protease generates amyloid-β (beta). At least 295 mutations in PSEN1 have been identified in AD. Pathogenic mutations of APP, a transmembrane protein, result in altered processing of the protein by secretases that leads to an increase in amyloid-beta production and/or an alteration in the ratio of specific amyloid-beta peptides. At least 54 mutations of the APP gene have been identified in AD. Another presenilin gene, PSEN2, which encodes for presenilin 2, is also a subunit of gamma-secretase involved in amyloid-beta generation. However, unlike PSEN1, PSEN2 is rarely involved in the development of early-onset AD. Approximately 50 mutations of PSEN2 have been identified in AD.7
As mentioned previously, TREM2, a transmembrane receptor that modulates microglial activity and survival, is involved in the pathogenesis of LOFAD, frontotemporal lobe dementia (FTD), and PD, indicating the significant overlap and interrelatedness of NDDs. TREM2 is also associated with the development of Nasu-Hakola disease, a rare autosomal recessive early-onset dementia. At least 48 mutations in TREM2 have been reported in AD. MAPT (microtubule-associated protein tau) encodes for the microtubule-associated protein, tau. Tau promotes the assembly of tubulin into microtubules and may be involved in axonal transport.8 However, in AD, tau is unable to bind to the microtubules because of hyperphosphorylation, which leads to the formation of toxic oligomers and paired helical filaments. Pathogenic mutations in this gene lead to changes in microtubule assembly and/or the tendency for tau to aggregate, leading to the classic plaques and tangles seen in AD. There are at least 15 mutations in MAPT seen in AD.9
Tauopathies, which include progressive supranuclear palsy, corticobasal degeneration, Pick's disease, argyrophilic grain disease, and FTD with parkinsonism are characterized as the formation of insoluble tau filaments leading to neurodegeneration.10
Genetic Testing
Whereas PSEN1, APP, and PSEN2 are "determinant genes,"playing a direct role in the pathogenesis of AD, APOEε4 is a "risk gene." Predictive testing of the three determinant genes in asymptomatic family members can be performed to assess risk. If a positive family history of EOFAD is present, testing of symptomatic relatives should be considered regardless of age. However, APOEε4 genotyping is not useful as a predictive test because although having one or two copies of the allele may increase risk, being negative for the allele does not eliminate risk.7,11
In February 2019, the Alzheimer's Association disseminated a fact sheet regarding the utility of the home genetic testing kit 23 and Me Personal Genome Service Genetic Health Risk (GHR) test. It warned that while the kit evaluates for the presence of the APOEε4 allele, it does not do so for the three determinant genes, thereby limiting its utility. The position of the Alzheimer's Association is that it does not recommend genetic testing for Alzheimer's disease for the general population.11
Mechanisms of Drugs in Development
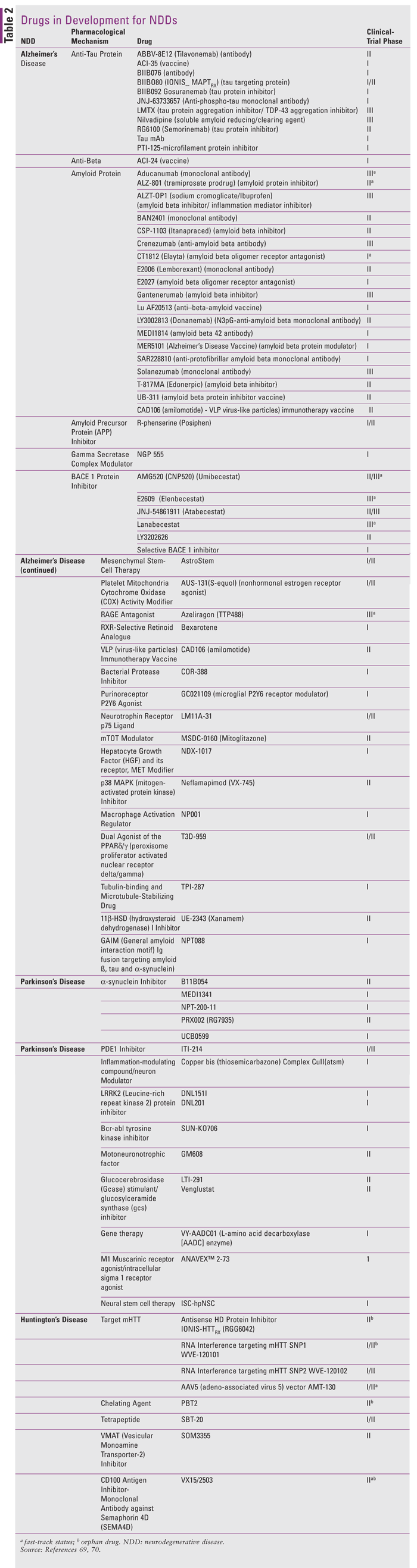
Principles of precision medicine, including targeting the underlying neuropathology, are being applied to numerous drugs that are currently in various phases of clinical trials for AD. Among the proposed mechanisms of drugs in development for AD are anti-tau and anti–beta-amyloid protein interventions; amyloid precursor protein (APP) inhibitors; gamma-secretase complex modulators; BACE (beta-secretase)-1 protein inhibitors; mesenchymal stem cell therapy, which may stimulate neurogenesis12; platelet mitochondria cytochrome oxidase activity modifiers—mitochondria dysfunction may underlie the development of AD13; RAGE (receptor for advanced glycation endproducts) antagonists—advanced glycation endproducts cause inflammation and oxidative damage14; RXR (retinoid X receptor)-selective retinoid analogues—impaired retinoic acid signaling results in neuroinflammation, oxidative stress, mitochondrial dysfunction, and neurodegeneration15,16; bacterial protease inhibitors of gingipains, cysteine proteases of the periodontal pathogen Porphyromonas gingivalis17; purinoreceptor P2Y6 agonists, which may stimulate microglial phagocytosis and inhibit the release of inflammatory cytokines by microglia18; neurotrophin receptor p75 (p75NTR) ligands— p75NTR regulates amyloid-beta metabolism in the brain19; mTOT (mitochondrial target of thiazolidinedione insulin sensitizers) modulators—insulin sensitization modulates mitochondrial metabolism)20; HGF-MET (hepatocyte growth factor and protooncogene receptor tyrosine kinase) receptor modifiers—growth factor and its receptor regulates brain function including axonal growth21; p38 MAPK (mitogen-activated protein kinase) inhibitors—p38 MAPK is a stress kinase expressed in microglia and neurons that releases proinflammatory cytokines22; macrophage activation regulators, which act as immune regulators of inflammatory monocytes/macrophages involved in neuroinflammation23; dual agonists of the PPARdelta/gamma (peroxisome proliferator activated nuclear receptor delta/gamma) which improves insulin sensitivity that may improve metabolic status24; tubulin-binding and microtubule-stabilizer25; 11 beta-HSD (hydroxysteroid dehydrogenase) I inhibitors, which inhibit an enzyme involved in stress response that is linked to amyloid plaque and neurofibrillary pathology26; and GAIM (general amyloid interaction motif) Ig fusion therapies, which target amyloid beta, tau, and alpha-synuclein.27 TABLE 2 identifies various genomic-based mechanisms of action for drugs currently undergoing clinical trials for AD, PD, and HD.
PARKINSON'S DISEASE
PD is an NDD that is characterized by tremor, rigidity, and bradykinesis and is due to loss of dopaminergic neurons in the substantia nigra with the finding of alpha-synuclein–containing Lewy bodies (which are intracytoplasmic inclusion bodies) in the remaining intact nigral neurons.28,29 Dementia and psychosis occur in 30% to 40% of patients as the disease progresses.28 The term parkinsonism encompasses PD, Lewy body dementia (LBD), FTD, drug-induced parkinsonism, Parkinson-plus diseases (i.e., multiplesystem atrophy, progressive supranuclear palsy, corticobasal degeneration) and others (i.e., autoimmune disease, vascular disease, viral/prion disease, Wilson's disease, normal-pressure hydrocephalus, heavy-metal exposure, dopa-responsive dystonia, and spinocerebellar ataxia).28
Approximately 1 million people in the U.S. have PD.30 Each year about 60,000 Americans are newly diagnosed.31 Epidemiologically, there are three cohorts of PD patients: juvenile-onset PD (JOPD), with onset occurring before age 20 years; early-onset adult PD (EOAPD), with onset between ages 20 and 50 years; and late-onset adult PD (LOAPD), with onset manifesting after the age of 50 years.28
PD can also be defined by the frequency of disease occurrence in families. This classification has recently been revised. PD typically occurs in only one family member; this was previously called idiopathic or sporadic PD. Currently, the preferred term is simplex case, meaning a PD patient with no family history for the disease. However, up to 15% of cases are associated with a positive family history for the disease. Five percent to 10% of PD is monogenic, that is, attributed to pathogenic variants in a single gene. Monogenic PD can be inherited in an autosomal dominant, autosomal recessive, or X-linked way. There are genes that are associated with EOAPD and LOAPD as well as JOPD. Typically, autosomal dominant disease manifests after age 50 years, whereas autosomal recessive disease appears before age 40 years. However, there is much interpatient variation between age of onset and gene expression (TABLE 1). Besides demonstrating the cardinal signs of PD, JOPD is also often associated with atypical features, including dystonia, spasticity, intellectual disability, seizures, hyperreflexia, oculomotor limitations, and cognitive loss.28
Although genetics explains a part of the etiology of PD, the interplay between genetics, age, and environment may play a larger role. As with AD, advancing age is the greatest risk factor for developing PD. Environmental toxins, including pesticide exposure, contribute to the development of PD. Other risk factors may include head injury, living in a rural area, or infectious diseases. Factors that may be protective against developing PD include cigarette smoking, caffeine ingestion, use of nonsteroidal anti-inflammatory drugs, hyperuricemia, and exercise.28
Alpha-synuclein is a presynaptic protein accounting for 1% of all cytosolic proteins.32 Normally, alpha-synuclein exists in the body in a soluble, unfolded state. In PD, there is a mutation in the SNCA gene that encodes for alpha-synuclein, resulting in misfolded neurotoxic forms of the protein that downregulate mitochondrial complex-1 activity, cause endoplasmic stress, disrupt cell-membrane integrity, inhibit the ubiquitin proteasome system, and impair autophagy. This aberrant protein accumulates intracellularly, resulting in Lewy pathology. Oligomers that form during aggregation produce oxidative stress, membrane penetrance, and synaptic and mitochondrial dysfunction leading to neuronal death.32-35 Recent evidence supports that the location of the initiation of misfolded forms of alpha-synuclein may extend beyond the central nervous system (CNS) to the olfactory bulb and enteric nervous system, which is part of the autonomic nervous system governing the gastrointestinal tract.36 As with tauopathies, synucleinopathies such as LBD and multiplesystem atrophy also occur and are due to insoluble alpha-synuclein filaments.10 Tau, along with alphasynuclein, amyloid-beta, and prion protein—all NDD-associated proteins—may be a driving force in NDDs.32
Genetic Testing
The recommendations for genetic testing for LOAPD of unknown case (formerly known as idiopathic or sporadic PD) are nonspecific since the disease is multifactorial, attributed to both environmental and genetic factors. PD caused by pathogenic variants of the genes GBA, LRRK2, SNCA2, or VPS35 results in autosomal dominant inheritance. Children of parents with any of these genetic mutations have a 50% chance of inheriting the pathogenic variant. The chances that the pathogenic variant will manifest increase with advancing age of the offspring. In the case of autosomal recessive inheritance, which is conferred by biallelic pathogenic variants of the genes ATP13A2, PARK7 (DJ-I), DNAJC6, FBOX07, PINK1, PODXL, PRKN, SLC6A3, SYNJ1, or VPS13C, children of an affected parent are obligate heterozygotes for the pathogenic variant. They may be at increased risk of developing PD. Predictive testing for the presence of the genes GBA, LRRK2, SNCA2, or VPS35 in asymptomatic individuals can be performed once the pathogenic variant has been identified in an affected family member. This testing, however, will not predict the age of onset of the disease.
Important issues to consider are psychological stress as well as possible stigma and societal discrimination that may result from the knowledge of one's genetic status. Testing of minors is not considered appropriate. Caution is advised when using direct-toconsumer genetic testing because such testing lacks pretest genetic counseling, personalization of possibly disturbing information, and supportive counseling should the result indicate an increased risk of developing PD.28
Mechanisms of Drugs in Development
As with AD, precision medicine in PD is targeting underlying genetic pathologies in the development of treatments for this NDD (TABLE 2).
In PD, focus has been placed on targeting toxic alphasynuclein. Proposed mechanisms of drugs currently in clinical trials include decreasing alpha-synuclein production by interfering with RNA, inhibiting alpha-synuclein aggregation, promoting intracellular breakdown of alpha-synuclein aggregates, enhancing lysosomal degradation, and promoting extracellular degradation of alpha-synuclein via immunization.37
Other mechanisms include inhibiting phosphodiesterase type 1 (PDE 1), which can increase cAMP and cGMP levels in the brain, enhancing energy stores that may be depleted38; correcting imbalances in copper homeostasis that are associated with the generation of reactive oxygen species and fibrillogenesis in PD39; inhibiting leucine-rich repeat kinase 2 (LRRK2), which plays a role in innate immunity involving both macrophages and microglia in the periphery and CNS40,41; blocking Bcr-abl tyrosine kinase, which is associated with the development of oxidative stress and is thought to play a role in PD induction42; acting as a motoneuronotrophic factor analog, which mimics an endogenous human embryonic-stage neural regulatory and signaling peptide43; inhibiting glucosylceramide synthase or stimulating glucocerebrosidase to increase levels of the lysosomal enzyme that is affected by mutations in the GBA gene in PD and Gaucher disease to help maintain basic cell function, correct lysosomal homeostasis, decrease endoplasmic reticulum stress, and improve mitochondrial activity44; developing gene therapy to deliver L-amino decarboxylase (AADC) enzyme, the enzyme that mediates the conversion of levodopa into dopamine, directly into the putamen area of the brain45; acting as a ligand for sigma-1 (an intraceullar chaperone)/ muscarinic receptors to enhance memory and confer neuroprotection by modulating inter-organelle signaling, attenuating endoplasmic reticulum stress and interacting with ion channels46,47; and the use of neural stem-cell therapy.48
HUNTINGTON'S DISEASE
HD is a monogenic, progressive NDD affecting motor, cognitive, and psychiatric disturbances caused by 36 or more trinucleotide repeats or expansions in the cytosine, adenine, guanine (CAG) sequence of the nucleotide. It is unique compared with many other NDDs in that the mutation exists in only one gene, the huntingtin gene, HTT. Mutations are then transcribed into messenger RNA leading to the production of abnormal RNA molecules and HTT protein. The disease is characterized by progressive motor disability including chorea, or rhythmic, snake-like movements, cognitive impairment, personality changes, depression, and a positive family history for the disease. The onset and severity of the disease are directly correlated with the number of trinucleotide repeats. However, in most patients the disease manifests between ages 35 to 44 years, and median survival is 15 to 18 years.49 It is estimated that more than 300,000 people in the U.S. have HD.50 There are both geographic and ethnic differences in the prevalence of HD, with Asians having the lowest rates and Caucasians having the highest rates.51 Interestingly, whereas cigarette smoking is protective in PD, in HD smoking—as well as levodopa and alcohol—may exacerbate the disease.49
HD is inherited in an autosomal dominant manner, so offspring of an affected parent have a 50% chance of inheriting the mutant allele. Allele size is directly correlated both to the risk of developing HD as well as the age of onset and age at death. A person having 26 or fewer CAG trinucleotide repeats is considered having normal alleles. The presence of 27 to 35 CAG trinucleotide repeats is considered having intermediate alleles because although the individual carrying this mutation is not at risk of developing HD, his/her offspring may be, because with each successive generation the number of repeats increases due to instability of the nucleotide. This process is called anticipation and refers to the phenomenon of increasing disease severity or decreasing age of onset in successive generations. CAG expansion is more common in paternally inherited intermediate alleles. Having 36 or more CAG trinucleotide repeats is considered having HD-causing alleles, with this further subdivided into reduced penetrance HD-causing alleles, i.e., those with 36 to 39 abnormal sequence repeats or fullpenetrance HD-causing alleles, i.e., those with 40 or more CAG trinucleotide repeats.
Although there is a chance that persons having the reduced penetrance alleles may not develop symptoms, the risk of developing HD is greatly increased in those with full-penetrance alleles. Larger CAG nucleotide sequences are more prone to expansion in future generations. Juvenile-onset HD is often associated with 60 or more CAG nucleotide repeats. Deterioration of motor, cognitive, and functional status is directly correlated to the number of repeat nucleotide sequences.49
Genetic Testing
Genetic testing consists of targeted analysis of the HTT gene, as 100% of probands (i.e., affected individuals) with the pathogenic HTT variant will be detected by testing this gene. Testing of asymptomatic at-risk individuals is not helpful for predicting age of onset, severity of the disease, symptomatology, or rate of progression, but it is useful in determining CAG repeat size, which would have bearing on the course of the disease should it develop. Testing is also beneficial if a carrier of mutant HTT (mHTT) is interested in being enrolled in a clinical trial.49
Mechanisms of Drugs in Development
Considering the underlying pathology of HD, drugs are being developed that target mHTT via various mechanisms, including chelation of metal-induced aggregation of mHTT52; the use of antisense HD protein inhibitors that target and destroy all forms of HTT, including normal HTT protein (as in the case of RG6042)53-55 or selectively target mHTT protein (as in the case of WVE-120101, which acts on the single nucleotide polymorphism 1 [SNP1] and WVE-120102, which acts on SNP2)56,57; or by the use of gene therapy to inhibit the production of mHTT.58,59 There is some concern with the nonspecific targeting of all HTT protein since it is thought that the normal protein is involved in proper neuronal functioning. It is still unclear what, if any, effect the nonselective silencing of HTT protein has on off-target sites.57,60,61 CRISPR is also being used to target toxic microsatellite repeat expansions of RNA (mHTT) in HD.62
Other neuroprotective mechanisms being studied among drugs in development for HD include targeting mitochondrial dysfunction in HD63,64; inhibiting vesicular monoamine transporter-2 (VMAT2), which reduces the amount of neurotransmitter stored in the vesicles, thereby decreasing the intensity of nerve signaling and choreic movements65; and using a monoclonal antibody to block the effects of SEMAD (Semaphorin 4D), a proton coding gene on microglia, and astrocyte activation, which is involved in neuroinflammation and neurodegeneration.66,67
Pharmacist's Role in Managing NDDs
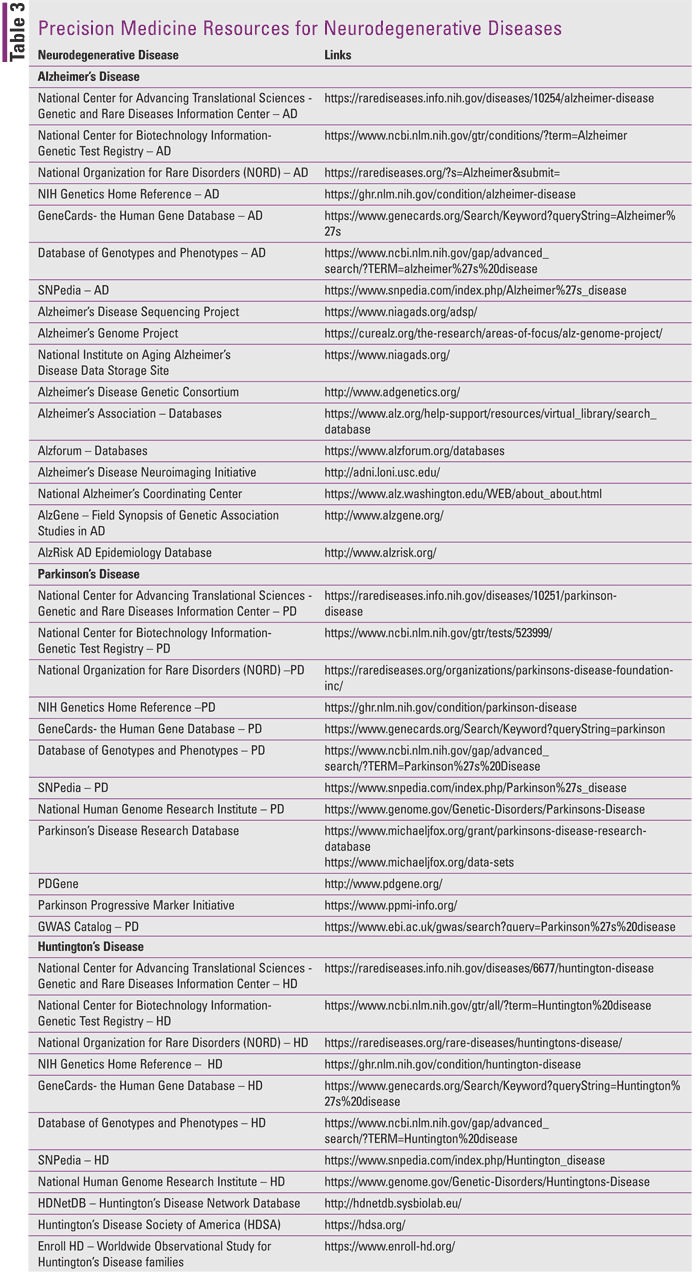
As the field of precision medicine evolves, it will continue to elucidate the mechanisms involved in the pathogenesis of NDDs. This is extremely important given that researchers can focus on developing medications that correct or reverse mutant variations that have been identified and are linked to disease expression. Pharmacists need to be knowledgeable about the underlying pathological processes involved in NDDs in order to communicate with both patients and other healthcare professionals. They also need to keep current with drugs in development and ongoing clinical trials that are available for their patients since there are no cures for these conditions. Professional resources are available to assist pharmacists in keeping up with the latest developments in this field (see TABLE 3, available online). Pharmacists can also offer recommendations for pharmacological interventions and emotionally supportive assistance to both patients and their caregivers as they navigate the difficulties of daily life in the face of declining physical and cognitive function and behavioral disturbances. The future holds great promise for finding effective and possibly curative treatments for these NDDs.
REFERENCES
- EU Joint Programme—Neurodegenerative Disease Research (JPND). What is neurodegenerative disease? www.neurodegenerationresearch.eu/ about/what/. Accessed November 10, 2019.
- Sheikh S, Safia, Haque E, Mir SS. Neurodegenerative diseases: multifactorial conformational diseases and their therapeutic interventions. J Neurodegener Dis. 2013;2013:563481.
- Dugger BN, Dickson DW. Pathology of neurodegenerative diseases. Cold Spring Harb Perspect Biol. 2017;9(7):a028035.
- Gan L, Cookson MR, Petrucelli L, et al. Converging pathways in neurodegeneration, from genetics to mechanisms. Nat Neurosci. 2018;21(10):1300-1309.
- Apostolova LG. Alzheimer disease. Continuum (Minneap Minn). 2016;22(2 Dementia):419-434.
- Alzheimer's Association. Alzheimer's disease facts and figures. 2019. www.alz.org/alzheimers-dementia/facts-figures. Accessed November 11, 2019.
- Bird TD. Alzheimer disease overview. October 23, 1998. Updated December 20, 2018. In: Adam MP, Ardinger HH, Pagon RA, et al., eds. GeneReviews [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2019. www.ncbi.nlm.nih.gov/books/NBK1161/. Accessed November 8, 2019.
- Stern JL, Lessard DV, Christopher RA, Berger L. Tau. Chapter 7. Single-molecule imaging of Tau dynamics on the microtubule surface. www. sciencedirect.com/topics/medicine-and-dentistry/tau. Accessed November 9, 2019.
- Alzforum.org. Mutations (Alzheimer's disease). www.alzforum.org/ mutations/search?genes=&diseases%5B%5D=145&keywordsentry=&keywords=#results. Accessed November 9, 2019.
- Siegel GJ, Albers RW, Price DL, et al. Basic Neurochemistry: Principles of Molecular, Cellular, and Medical Neurobiology, 8th edition. Academic Press; Amsterdam: 2012. Ebook.
- Alzheimer's Association. Genetic testing. TS-0044. February 2019. https://alz.org/media/Documents/alzheimers-dementia-genetic-testing-ts. pdf. Accessed November 9, 2019.
- Staff NP, Jones DT, Singer W. Mesenchymal stromal cell therapies for neurodegenerative diseases. Mayo Clin Proc. 2019;94(5):892-905.
- Cardoso SM, Proenca MT, Santos S, et al. Cytochrome c oxidase is decreased in Alzheimer's disease platelets. Neurobiol Aging. 2004;25:105-110.
- Alzforum Therapeutics. Azeliragon. www.alzforum.org/therapeutics/ azeliragon. Accessed November 5, 2019.
- Das BC, Dasgupta S, Ray SK. Potential therapeutic roles of retinoids for prevention of neuroinflammation and neurodegeneration in Alzheimer's disease. Neural Regen Res. 2019;14(11):1880-1892.
- Fitz NF, Nam KN, Koldamova R, et al. Therapeutic targeting of nuclear receptors, liver X and retinoid X receptors, for Alzheimer's disease. Br J Pharmacol. 2019;176(18):3599-3610.
- Alzforum Therapeutics. Cor388. www.alzforum.org/therapeutics/ cor388. Accessed November 8, 2019.
- Alzforum Therapeutics. GC 021109. www.alzforum.org/therapeutics/gc-021109. Accessed November 8, 2019.
- Xu Y, Li WW, Wang J, et al. Neurotrophin receptor p75 mRNA level in peripheral blood cells of patients with Alzheimer's disease. Neurotox Res. 2019;36(1):101-107.
- Shah RC, Matthews DC, Andrews RD, et al. An evaluation of MSDC-0160, a prototype mTOT modulating insulin sensitizer, in patients with mild Alzheimer's disease. Curr Alzheimer Res. 2014;11: 564-573.
- Inacio P. NDX-1017 phase I trial complete, results will be presented at CTAD conference, Athira says. October 9, 2019. https://alzheimersnewstoday.com/2019/10/09/ndx-1017-phase-1-trial-complete-results-willbe-presented-ctad-conference-athira-says/. Accessed November 8, 2019.
- Alzforum Therapeutics. Neflamapimod. www.alzforum.org/therapeutics/neflamapimod. Accessed November 8, 2019.
- Centerwatch. NP001 Alzheimer's disease and blood markers of inflammation. www.centerwatch.com/clinical-trials/listings/120240/ alzheimer-disease-np001-alzheimers-disease/. Accessed November 8, 2019.
- Alzforum Therapeutics. T3D-959. www.alzforum.org/therapeutics/ t3d-959. Accessed November 8, 2019.
- Mondal P, Das G, Khan J, et al. Crafting of neuroprotective octapeptide from taxol-binding pocket of β-tubulin. ACS Chem Neurosci. 2018;9(3):615-625.
- Alzforum Therapeutics. XanamemTM. www.alzforum.org/therapeutics/xanamemtm. Accessed November 8, 2019.
- Michelson D, Grundman M, Magnuson K, et al. Randomized, placebo-controlled trial of NPT088, a phage-derived, amyloid-targeted treatment for Alzheimer's Disease. J Prev Alzheimers Dis. 2019;6(4):228-231.
- Cook Shukla L, Schulze J, Farlow J, et al. Parkinson disease overview. May 25, 2004. Updated July 25, 2019. In: Adam MP, Ardinger HH, Pagon RA, et al., eds. GeneReviews [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2019. www.ncbi.nlm.nih.gov/books/ NBK1223/. Accessed November 8, 2019.
- Postuma RB, Berg D, Stern M, et al. MDS clinical diagnostic criteria for Parkinson's disease. Movement Dis. 2015;30:1591-1598.
- Parkinson's News Today. Parkinson's Disease statistics. https://parkinsonsnewstoday.com/parkinsons-disease-statistics/. Accessed November 7, 2019.
- Parkinson's Foundation. Statistics. www.parkinson.org/Understanding-Parkinsons/Statistics. Accessed November 7, 2019.
- Visanji NP, Lang AE, Kovacs GG. Beyond the synucleinopathies: alpha synuclein as a driving force in neurodegenerative comorbidities. Transl Neurodegener. 2019;8:28.
- Killinger BA, Kordower JH. Spreading of alpha-synuclein-relevant or epiphenomenon? J Neurochem. 2019;150(5):605-611.
- Stefanis L. α-Synuclein in Parkinson's disease. Cold Spring Harb Perspect Med. 2012;2(2):a009399.
- Xu MM, Ryan P, Rudrawar S, et al. Advances in the development of imaging probes and aggregation inhibitors for alpha-synuclein. Acta Pharmacol Sin. October 4, 2019. Epub ahead of print. www.ncbi.nlm. nih.gov/pubmed/31586134.
- Mazurskyy A, Howitt J. Initiation and transmission of alpha-synuclein pathology in Parkinson's disease. Neurochem Res. November 11, 2019. Accessed November 9, 2019.
- Savitt D, Jankovic J. Targeting alpha-synuclein in Parkinson's disease: progress towards the development of disease-modifying treatments. Drugs. 2019;79(8):797-810.
- Medina AE. Therapeutic utility of phosphodiesterase type 1 inhibitors in neurological conditions. Front Neurosci. 2011;5:21.
- Ilyechova EY, Miliukhina IV, Orlov IA, et al. A low blood copper concentration is a co-morbidity burden factor in Parkinson's disease development. Neuroscience Res. 2018;135:54-62.
- Lee H, James WS, Cowley SA. LRRK2 in peripheral and central nervous system innate immunity: its link to Parkinson's disease. Biochem Soc Trans. 2017;45(1):131-139.
- Dagan E, Schlesinger I, Kurolap A, et al. LRRK2, GBA and SMPD1 founder mutations and Parkinson's disease in Ashkenazi Jews. Dement Geriatr Cogn Disord. 2016;42(1-2):1-6.
- Brahmachari S, Karuppagounder SS, Ge P, et al. c-Abl and Parkinson disease: mechanisms and therapeutic potential. J Parkinsons Dis. 2017;7(4):589-601.
- Good Clinical Practice Network. ICH GGP I Clinical Trials Registry. GM 608 in a phase IIA pilot double-blinded, randomized, placebo controlled trial in mild to moderate Parkinson disease phase 2a study of gm 608 in mild to moderate Parkinson disease. https://ichgcp.net/clinical-trials-registry/NCT01850381. Accessed November 5, 2019.
- Do J, McKinney C, Sharma P, et al. Glucocerebrosidase and its relevance to Parkinson disease. Mol Neurodegener. 2019;14:36.
- Inacio P. Parkinson's News Today. #AANAM-Investigational VYAADC01 gene therapy provides benefits for Parkinson's patients, Phase I data show. May 7, 2019. https://parkinsonsnewstoday.com/2019/05/07/ aanam-vy-aadc01-provides-benefits-parkinsons-patients-phase-1-trial/. Accessed November 5, 2019.
- Alzform Therapeutics. Anavex 2-73. www.alzforum.org/therapeutics/ anavex-2-73. Accessed November 5, 2019.
- Penke B, Fulop L, Szucs M, et al. The role of sigma-1 receptor, an intracellular chaperone in neurodegenerative diseases. Curr Neuropharmacol. 2018;16:97-116.
- Garitaonandia I, Gonzalez R, Sherman G, et al. Novel approach to stem cell therapy in Parkinson's disease. Stem Cells Dev. 2018. 27(14):951-957.
- Caron NS, Wright GEB, Hayden MR. Huntington disease. October 23, 1998. Updated July 5, 2018. In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2019. www.ncbi.nlm.nih.gov/books/ NBK1305/ Accessed November 5, 2019.
- National Institute of Neurological Diseases and Stroke. Huntington's disease. www.ninds.nih.gov/Disorders/All-Disorders/Huntingtons-Disease-Information-Page. Accessed November 9, 2019.
- Rawlins MD, Wexler NS, Wexler AR, et al. The prevalence of Huntington's disease. Neuroepidemiology. 2016;46(2):144-153.
- Huntington Study Group Reach2HD Investigators. Safety, tolerability, and efficacy of PBT2 in Huntington's disease: a phase 2, randomised, double-blind, placebo-controlled trial. Lancet Neurol. 2015;14(1):39-47.
- Tabrizi SJ, Leavitt BR, Landwehrmeyer GB, et al for the Phase 1-2a IONIS-HTTRx Study Site Teams. Targeting huntingtin expression in patients with Huntington's disease. N Engl J Med. 2019;380(24):23072316.
- Lopes JM. Huntington's Disease News. Phase 3 trial of RG6042 will gauge potential of gene-silencing approaches for Huntington's, other diseases. February 5, 2019. https://huntingtonsdiseasenews.com/2019/02/05/ phase-3-trial-rg6042-will-inform-potential-gene-silencing-huntingtonsother-neurodegenerative-diseases/. Accessed November 5, 2019.
- Inacio P. Huntington's Disease News. Investigational RG6042 lowers mutant huntingtin protein in early-stage patients, phase 1/2 data show. June 4, 2019. https://huntingtonsdiseasenews.com/2019/06/04/inionishttrx-decreases-mutant-huntingtin-protein-early-stage-huntingtons/. Accessed November 8, 2019.
- Hersch S, Claassen D, Edmondson M, et al. Multicenter, randomized, double-blind, placebo-controlled phase 1a/2b studies of WVE120101 and WVE-120102 in patients with Huntington's disease. Neurology. 2017;88 (16 Supplement).
- Huntington's Disease News. WVE-120102. https://huntingtonsdiseasenews.com/wve-120102/. Accessed November 8, 2019.
- Huntington's Disease News. AMT-130. https://huntingtonsdiseasenews.com/amt-130/. Accessed November 8, 2019.
- Inacio P. Huntington's Disease News. FDA places gene therapy AMT-130 on fast track to speed development. April 11, 2019. https:// huntingtonsdiseasenews.com/2019/04/11/fda-places-gene-therapy-amt130-on-fast-track-to-speed-development/. Accessed November 8, 2019.
- Huntington Study Group. Meet the compounds: WVE-120101 and WVE-120102. https://huntingtonstudygroup.org/hd-insights-category/ meet-the-compounds-wve-120101-and-wve-120102/. Accessed November 8, 2019.
- Globe Newswire. Wave Live Sciences provides timing update on PRECISION-HD clinical programs. www.globenewswire.com/newsrelease/2019/04/10/1801872/0/en/Wave-Life-Sciences-Provides-TimingUpdate-on-PRECISION-HD-Clinical-Programs.html. Accessed November 8, 2019.
- Batra R, Nelles DA, Pirie E, et al. Elimination of toxic microsatellite repeat expansion RNA by RNA-targeting Cas9. Cell. 2017;170(5):899-912.
- Huntington's Disease News. SBT-20. https://huntingtonsdiseasenews.com/sbt-20/. Accessed November 8, 2019.
- Stealth BioTherapeutics. Stealth BioTherapeutics initiates Phase 1/2 trial of SBT-20 in patients with early stage Huntington's disease. https:// www.prnewswire.com/news-releases/stealth-biotherapeutics-initiatesphase-12-trial-of-sbt-20-in-patients-with-early-stage-huntingtons-disease-300449156.html. Accessed November 8, 2019.
- Huntington's Disease News. SOM3355. https://huntingtonsdiseasenews.com/som3355/. Accessed November 8, 2019.
- Huntington's Disease News. VX15/2503. https://huntingtonsdiseasenews.com/vx152503/. Accessed November 8, 2019.
- Globe Newswire. Vaccinex, Inc. announces completion of enrollment for its "SIGNAL" Huntington's disease trial. https://www.globenewswire.com/news-release/2019/01/16/1696711/0/en/Vaccinex-IncAnnounces-Completion-of-Enrollment-for-Its-SIGNAL-Huntington-s-Disease-Trial.html. Accessed November 8, 2019.
- National Center for Biotechnology Information, U.S. National Library of Medicine-ClinVar. https://www.ncbi.nlm.nih.gov/clinvar/. Accessed November 1, 2019.
- Pharmaceutical Research and Manufacturing Association. Medicines in development for neurological disorders 2018. www.phrma.org/-/media/Project/PhRMA/PhRMA-Org/PhRMA-Org/PDF/MID_Neurological-Disorders-Drug-List_2018.pdf. Accessed November 1, 2019.
- Adis Insights. https://adisinsight.springer.com/. Accessed November 1, 2019.