Psychotropic Drug Interactions: A Review
RELEASE DATE:
November 1, 2017
EXPIRATION DATE:
November 30, 2019
FACULTY:
Emily K. Sochovka, PharmD Candidate 2018
John G. Riles, PharmD Candidate 2018
Crystal M. Deas, PharmD, BCPS
Assistant Professor of Pharmacy Practice
Jeffrey A. Kyle, PharmD, BCPS
Professor of Pharmacy Practice
Samford University McWhorter School of Pharmacy
Birmingham, Alabama
FACULTY DISCLOSURE STATEMENTS:
Drs. Deas and Kyle, Ms. Sochovka, and Mr. Riles have no actual or potential conflicts of interest in relation to this activity.
Postgraduate Healthcare Education, LLC does not view the existence of relationships as an implication of bias or that the value of the material is decreased. The content of the activity was planned to be balanced, objective, and scientifically rigorous. Occasionally, authors may express opinions that represent their own viewpoint. Conclusions drawn by participants should be derived from objective analysis of scientific data.
ACCREDITATION STATEMENT:
Pharmacy
 Postgraduate Healthcare Education, LLC is accredited by the Accreditation Council for Pharmacy Education as a provider of continuing pharmacy education.
Postgraduate Healthcare Education, LLC is accredited by the Accreditation Council for Pharmacy Education as a provider of continuing pharmacy education.
UAN: 0430-0000-17-076-H01-P
Credits: 2.0 hours (0.20 ceu)
Type of Activity: Knowledge
TARGET AUDIENCE:
This accredited activity is targeted to pharmacists. Estimated time to complete this activity is 120 minutes.
Exam processing and other inquiries to:
CE Customer Service: (800) 825-4696 or cecustomerservice@jobson.com
DISCLAIMER:
Participants have an implied responsibility to use the newly acquired information to enhance patient outcomes and their own professional development. The information presented in this activity is not meant to serve as a guideline for patient management. Any procedures, medications, or other courses of diagnosis or treatment discussed or suggested in this activity should not be used by clinicians without evaluation of their patients' conditions and possible contraindications or dangers in use, review of any applicable manufacturer's product information, and comparison with recommendations of other authorities.
GOAL:
To provide an overview of the common pathways of psychotropic drug interactions and to discuss the significance of these interactions, as well as strategies for avoiding patient harm.
OBJECTIVES:
After completing this activity, the participant should be able to:
- Differentiate between pharmacokinetic and pharmacodynamic drug interactions.
- Describe pharmacokinetic drug-interaction pathways that may influence the safety and efficacy of antidepressants, anxiolytics, and atypical antipsychotics.
- Identify clinically significant consequences of pharmacodynamic interactions involving antidepressants, anxiolytics, and atypical antipsychotics.
- Discuss strategies pharmacists may use to prevent psychotropic drug interactions.
ABSTRACT: Psychotropic drugs account for approximately one-fourth of the top 200 prescribed-drug sales in the United States and are implicated in a number of drug interactions. Mental illness is associated with an increased occurrence of chronic conditions; for this reason, many people with psychiatric disorders potentially could be on multiple medication regimens. Clinically significant drug interactions are events in which a drug’s pharmacodynamic (PD) or pharmacokinetic (PK) characteristics are modified by the addition of a second drug to the patient’s medication regimen. Most interactions involve PK pathways via CYP metabolism, and PD interactions are often more complex. Pharmacists are uniquely positioned to assist providers in the identification and resolution of interactions involving psychotropic drug therapy and to employ strategies to prevent patient harm.
It is estimated that more than 40 million Americans have a mental-health condition.1 According to a recent study, more than one in six persons older than 18 years of age are taking a psychotropic drug.2 Furthermore, mental illness is associated with an increased occurrence of chronic conditions such as cardiovascular disease, diabetes, obesity, asthma, epilepsy, and cancer.3 For this reason, many people with psychiatric disorders potentially could be on multiple medication regimens. As the number of a patient’s medications increases, so does the possibility of adverse drug reactions (ADRs), particularly those resulting from drug interactions.
Drug interactions vary in validity. The overall prevalence of drug interactions is estimated to be 50% to 60%.4 Some interactions are well documented, and others are anecdotal; in some cases, a drug may not have any clinically significant effects. Clinically significant drug interactions are events in which the pharmacodynamic (PD) or pharmacokinetic (PK) characteristics of a drug are modified when a second drug is added to the patient’s medication regimen. This alteration may result in serious adverse reactions or attenuation of efficacy.5,6 Interactions that affect pharmacodynamics or pharmacokinetics have a prevalence of approximately 5% to 9%.4 PK interactions, which are more common, involve alterations in absorption, distribution, metabolism, or excretion of a primary drug caused by the addition of a second drug with altering properties.7 Essentially, a PK interaction is what the body does to the drug; by contrast, a PD interaction is a consequence of what the drug does to the body. Additive or antagonistic effects caused by the coadministration of drugs with similar target sites of action are classified as PD interactions.8,9
Psychotropic drugs account for approximately one-fourth of the top 200 prescribed-drug sales in the United States and are implicated in a number of drug interactions.10 For pharmacists, identifying and preventing both known and potential interactions is crucial for proper management, optimization of drug therapy, and prevention of significant ADRs. This lesson provides an overview of common pathways of psychotropic drug interactions and discusses these interactions’ clinical significance, as well as strategies for avoiding patient harm.
OVERVIEW OF PK-INTERACTION PATHWAYS
As discussed above, PK interactions are caused by alterations in an agent’s absorption, metabolism, distribution, or elimination pathway.7 PK modifications of a drug’s action may be agonistic or antagonistic, or they may have no significant effect.11
Metabolism: Enzyme Induction and Inhibition
Alterations in drug metabolism resulting from enzyme induction and inhibition are commonly implicated in PK interactions. Most of these interactions involve the process of phase I (oxidative) metabolism, which is mediated by hepatic CYP450 enzymes. Numerous CYP enzymes have been identified (TABLE 1); however, six core enzymes—CYP1A2, CYP2B6, CYP2C9, CYP2C19, CYP2D6, and CYP3A4—are responsible for approximately 90% of all CYP450 activity.12 Coadministration of CYP substrates and drugs with activity on these core enzymes can impact therapeutic efficacy and the risk of drug toxicity. CYP3A4, the primary enzyme implicated in drug interactions, is involved in the metabolism of more than 50% of prescribed drugs.13 Induction of CYP activity results in increased substrate metabolism and may decrease drug efficacy, whereas inhibition may lead to drug accumulation, increased risk of ADRs, and potentially reduced drug exposure in cases of impaired activation of prodrugs. Several drugs are CYP substrates and may induce or inhibit the activity of various CYP enzymes. Likewise, some common foods (e.g., grapefruit juice) contain CYP inhibitors and inducers that may influence drug metabolism.
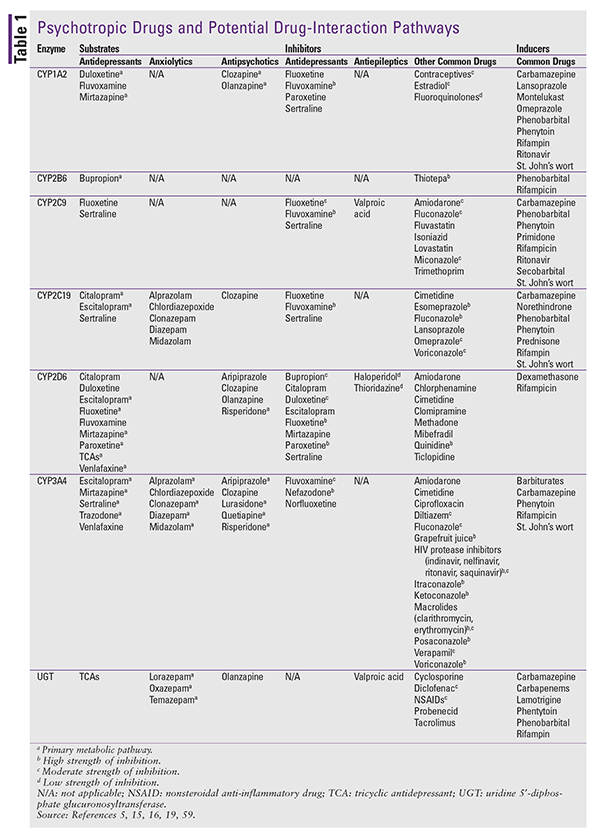
Phase II (conjugation) metabolism, which is mediated by the uridine 5'-diphosphate glucuronosyltransferase (UGT) family of enzymes, is another potential pathway for drug interactions.13 Phase II follows phase I oxidation and is characterized by the transformation of drug molecules to more hydrophilic substances that are more readily excreted. The enzymes involved in phase II function primarily to facilitate drug elimination and therefore play a smaller role in drug interactions compared with phase I enzymes.13 Although they are less significant than phase I–mediated interactions, UGTs may alter the therapeutic efficacy and influence the toxicity of agents that undergo significant metabolism via this pathway (e.g., olanzapine, amitriptyline).
P-Glycoprotein
P-glycoprotein (Pgp) is a transmembrane protein with several functions. It is involved in preventing intestinal absorption of certain drugs, promoting drug excretion by the liver and kidneys, and reducing tissue accumulation of various agents.14 Numerous tissues express Pgp, and Pgp activity is mediated by factors including genetic polymorphisms and specific drugs (e.g., verapamil). Agents that inhibit intestinal Pgp may enhance the bioavailability of Pgp substrates, thereby increasing the risk of toxicity, whereas inducers of Pgp produce lower drug concentrations of substrates in the body.
Absorption, Distribution, and Elimination
Disruptions in drug absorption, distribution, and elimination present additional pathways for PK interactions. Physiological changes at the absorption site resulting from alterations of gastric pH and reductions in drug solubility are mechanisms of PK pathways that affect drug absorption. Drug distribution is impacted primarily by disturbances in protein binding, and elimination is affected mainly by renal and hepatic dysfunction.13 Any combination of PK pathways may be involved in the occurrence of drug interactions with psychotropic drugs.
PK-INTERACTION PATHWAYS OF PSYCHOTROPIC DRUG CLASSES
Antidepressants
Selective Serotonin Reuptake Inhibitors: Selective serotonin reuptake inhibitors (SSRIs) are the most frequently prescribed antidepressants, and their use is widespread because of their safety profile, tolerability, selective mechanisms, and relative safety in the elderly population. Additionally, compared with other antidepressants, SSRIs appear to have a lower potential for drug interactions, with a few notable exceptions.15 The most common drug interactions encountered with this class are due to PK interaction via CYP450 enzymes, and each SSRI varies with respect to CYP450 activity.16
Both fluoxetine and paroxetine are predominantly metabolized by CYP2D6, and both are potent CYP2D6 inhibitors.15,16 Fluoxetine also moderately inhibits CYP2C9, CYP2C19, and CYP3A4. Apart from being a potent CYP2D6 inhibitor, paroxetine only mildly inhibits other CYP450 enzymes.16 Caution should be used in combining these drugs with CYP2D6 substrates, such as aripiprazole, carvedilol, clozapine, donepezil, galantamine, metoprolol, risperidone, and tricyclic antidepressants (TCAs) because increased drug levels and toxicities may occur.15,16 In addition, the inhibitory effects of paroxetine and, possibly, fluoxetine have been implicated in the reduction of tamoxifen’s efficacy through inhibition of that drug’s conversion to its active metabolite. This potential interaction could increase the risk of breast-cancer treatment failure and recurrence.15
Sertraline is predominantly metabolized by CYP3A4 and is categorized as a weak inhibitor of CYP450 enzymes.15,16 Therefore, this agent has a low PK drug-interaction potential, with one possible exception: Occasional case reports have demonstrated a significant interaction between sertraline and clozapine in that increased clozapine plasma levels were associated with increased doses of sertraline. Therefore, caution is advised when these agents are used concomitantly.15
Citalopram and escitalopram are weak CYP inhibitors and pose minimal risk of PK interactions.15,16 Both CYP2D6 and CYP2C19 play a role in the biotransformation and metabolism of citalopram to escitalopram; therefore, alterations in the activity of these enzymes may impact therapeutic efficacy.16 Although citalopram and escitalopram are unlikely to cause significant PK interactions, they should be used with caution with QTc-prolonging drugs such as TCAs and antipsychotics.15
Serotonin-Norepinephrine Reuptake Inhibitors: Serotonin-norepinephrine reuptake inhibitors (SNRIs) act as serotonin reuptake inhibitors at lower doses and as norepinephrine reuptake inhibitors at higher doses.15 Venlafaxine and duloxetine are the most commonly prescribed agents in this class, and these drugs differ with regard to their potential for interaction.
Venlafaxine is predominantly metabolized by CYP2D6 to the active metabolite desvenlafaxine, with CYP3A4 metabolism involved as a secondary process.15,17 Given the role of CYP3A4 and CYP2D6 in venlafaxine metabolism, caution should be exercised with combination therapy using venlafaxine and inducers or inhibitors of these enzymes. Commonly prescribed CYP2D6 inhibitors include diphenhydramine, terbinafine, bupropion, fluoxetine, and paroxetine, which may increase plasma levels of venlafaxine, leading to an increased risk of venlafaxine adverse effects.15,18 Because both venlafaxine and its active metabolite, desvenlafaxine, exert SNRI activity, CYP2D6 inhibitors may cause an increase in venlafaxine adverse effects and SNRI activity. CYP2D6 metabolism in vivo is generally thought to be noninducible.
Duloxetine is extensively metabolized via CYP1A2, with minor involvement of CYP2D6. Cigarette smoking induces CYP1A2 activity, resulting in decreased plasma drug concentrations. Therefore, patients who smoke may require higher doses of duloxetine to achieve desired therapeutic effects. Duloxetine is a moderate CYP2D6 inhibitor, so caution should be exercised in combining duloxetine with CYP2D6 substrates because duloxetine might increase plasma levels of those medications.15 Duloxetine has also been noted to increase risperidone concentrations by a mean of 26% via the CYP2D6 pathway and is linked to an increased risk of adverse effects associated with this agent when used in combination.19 Additionally, studies have indicated that duloxetine may impact warfarin therapy and increase international normalized ratio (INR) values because of the displacement of warfarin from its protein-binding site. INR should be monitored closely if these drugs are administered concurrently. Fluvoxamine, an SSRI, has been shown in some studies to increase the area under the time/plasma concentration curve of duloxetine by about 460%; therefore, caution should be used with coadministration.15,19
Serotonin Antagonist and Reuptake Inhibitor: Trazodone is a commonly prescribed serotonin antagonist and reuptake inhibitor. It is metabolized mainly by CYP3A4, and caution should be used with CYP3A4 inhibitors and inducers.15 Trazodone does not have a significant impact on the metabolism of other medications.16
TCAs: TCAs are subject to many PK interactions owing to their drug metabolism and narrow therapeutic index. They are metabolized primarily via CYP2D6, with CYP1A2, CYP3A4, and CYP2C19 involved as secondary pathways.16 Given these characteristics, TCAs are significantly impacted by inducers and inhibitors of CYP2D6, CYP1A2, and CYP3A4.15,20 Inhibition of these CYP enzymes may increase plasma levels of TCAs, which in turn could result in serious ADRs, such as arrhythmias, convulsions, confusion, or anticholinergic effects. These ADRs are concentration-dependent and are more problematic in vulnerable populations, such as the elderly.16 Whereas certain CYP450 inhibitors may cause an increase in plasma TCA concentrations, CYP450 inducers may decrease plasma TCA levels, leading to reduced efficacy.
Most TCAs are weak CYP450 inhibitors and inducers and do not affect the elimination of other drugs. In terms of the potential to cause PK interactions, desipramine and nortriptyline are the least problematic TCAs, as studies have shown that they are weak CYP2D6 inhibitors. However, the tertiary amine TCAs (amitriptyline, imipramine, clomipramine, and doxepin) are more potent CYP450 inhibitors and are more likely to be involved in clinically significant drug interactions.21
Norepinephrine-Dopamine Reuptake Inhibitor: Bupropion, a norepinephrine-dopamine reuptake inhibitor, is metabolized predominantly by CYP2B6.5 Bupropion is also categorized as a moderate-to-potent CYP2D6 inhibitor and has demonstrated metabolic effects with venlafaxine and other CYP2D6 substrates when administered concurrently.5,22 Bupropion may lower the seizure threshold at higher doses; therefore, agents that inhibit its metabolism should be used with caution and the patient should be carefully monitored.
Noradrenergic and Specific Serotonergic Antagonist: Mirtazapine, a noradrenergic and specific serotonergic antagonist, has three main metabolic pathways via CYP2D6, CYP1A2, and CYP3A4.23 Its metabolism through multiple routes suggests that mirtazapine has a low potential for PK interactions because alternative metabolic routes are available in situations in which the main route of metabolism is inhibited. Studies indicate that mirtazapine is a weak inhibitor of various CYP enzymes and suggest that it has a low PK-interaction potential.23
Anxiolytics
Benzodiazepines (BZDs) have various routes of metabolism. Lorazepam, oxazepam, and temazepam are devoid of CYP interactions and are metabolized primarily by UGTs, whereas alprazolam, clonazepam, diazepam, triazolam, and midazolam require metabolic oxidation through CYP3A4 and CYP2C19 prior to conjugation. The antidepressants fluoxetine, fluvoxamine, and paroxetine have been shown to increase serum concentrations of BZDs by 30% to 100% when used concomitantly, owing to CYP2C19 inhibition.24-26 Other antidepressants, such as sertraline, citalopram, escitalopram, venlafaxine, mirtazapine, and duloxetine, may be safely administered with BZDs, as they have limited impact on BZD metabolism. Additionally, the CYP3A4 inducers rifampin, phenytoin, and hypericum (St. John’s wort) can increase metabolism and decrease the serum concentration of BZDs metabolized through this pathway. Increased plasma concentrations of BZDs can occur when a BZD is coadministered with 3A4 inhibitors such as cimetidine, omeprazole, azole antifungals, and macrolides. Whereas lorazepam, oxazepam, and temazepam are less susceptible to CYP PK interactions, UGT inhibitors and inducers may affect drug therapy. The most common UGT inhibitors that can interfere with the therapeutic efficacy of BZDs are nonsteroidal anti-inflammatory drugs, immunosuppressants, valproic acid, and diclofenac. Some CYP3A4 inhibitors or inducers are also known to exert similar effects on UGT.27 Clinicians should monitor patients for signs and symptoms of BZD toxicity (e.g., mental status, dizziness, ataxia, respiratory depression) when an interaction is possible.
Buspirone’s mechanism of action is not understood; however, the drug possesses anxiolytic properties and is an attractive option for prescribers because of its ability to modulate serotonin and dopaminergic receptors without sedative and other neurologic adverse effects that commonly occur with BZDs. Buspirone is metabolized primarily via oxidative reduction mediated by CYP3A4, making it susceptible to PK interactions. Significant increases in plasma concentrations have been demonstrated with coadministration of buspirone and diltiazem, verapamil, erythromycin, itraconazole, nefazodone, and grapefruit juice.28 Reductions in plasma concentrations have been documented when buspirone is used concomitantly with rifampin. CYP3A4 inhibitors and inducers may affect bupropion metabolism and should be used with caution.28
Atypical Antipsychotics
Most atypical antipsychotics do not have significant activity on CYP or Pgp enzymes. However, several agents are CYP substrates and are vulnerable to PK interactions. Clozapine is metabolized primarily by CYP1A2 and secondarily by CYP2C19, CYP3A4, and CYP2D6. Clozapine’s narrow therapeutic index and increased potential for drug interactions and consequent side effects means that patients must undergo routine drug-level and toxicity monitoring. Agents that inhibit CYP metabolism of clozapine result in increased plasma concentrations that may decrease the seizure threshold and increase the risk of hematologic toxicity. The decision to coadminister clozapine and CYP1A2 inhibitors should be considered carefully.
Fluvoxamine, an inhibitor of CYP1A2 and CYP2C19 metabolic pathways, is reported to cause a five- to 10-fold increase in plasma clozapine levels with concomitant therapy.29-36 Other notable CYP1A2 inhibitors that may increase clozapine plasma concentrations are ciprofloxacin and cimetidine. In general, antiepileptic drugs are inducers of one or more enzymes involved in clozapine metabolism. Carbamazepine, phenytoin, and phenobarbital are strong CYP1A2 and CYP3A4 inducers and should be avoided in combination with clozapine because of the risk of ADRs. If combination therapy cannot be avoided, clozapine doses, along with the patient’s clinical status, should be monitored closely. CYP1A2 induction resulting from smoking has been shown to reduce clozapine plasma levels, and regimen adjustments may be required based on therapeutic drug monitoring.
Olanzapine is metabolized primarily by CYP1A2 and secondarily metabolized by CYP2D6 and UGT. Its PK-interaction potential is similar to that of clozapine via CYP1A2 inhibition and induction.37-40 Risperidone is metabolized primarily via CYP2D6 and secondarily by CYP3A4. Therefore, concomitant use of risperidone with CYP2D6 inhibitors and inducers of risperidone metabolism should be approached with caution. Quetiapine is metabolized primarily by CYP3A4, and caution should be exercised when it is used with potent inhibitors and inducers of this enzyme.
The newer antipsychotics, lurasidone and aripiprazole, have fewer documented PK interactions. Lurasidone is metabolized primarily by CYP3A4, which results in two pharmacologically active metabolites that may pose clinical challenges if lurasidone metabolism is altered. Notably, coadministration of lurasidone and ketoconazole is contraindicated because of an almost 10-times increase in AUC.41 Concomitant use of lurasidone and moderate CYP3A4 inhibitors is appropriate provided that the dosage does not exceed 40 mg per day. Aripiprazole is metabolized primarily via CYP2D6 and CYP3A4. CYP3A4 inducers such as rifampin can dramatically increase aripiprazole clearance. Dose adjustments of aripiprazole are recommended upon initiation or discontinuation of strong CYP3A4 inducers.42
OVERVIEW OF PD-INTERACTION PATHWAYS
PD interactions occur when an agent alters the response of another drug, resulting in synergism or antagonism of clinical effects.8,9 Synergism may be beneficial if it is used to enhance therapeutic efficacy, such as in a situation of combination psychotropic medications used to manage a treatment-resistant form of mental illness.8,9 Conversely, additive drug toxicity from synergism represents an undesired PD interaction. Similarly, antagonistic PD interactions may attenuate or enhance drug effects and influence patient outcomes. Because of an overlap in drug therapy targets and similarities in side-effect profiles, the risk of PD interactions is increased with concomitant psychotropic medications. Serotonin syndrome, hypotension, increased fall risk, and hematologic effects are some of the clinical consequences of potentially harmful PD interactions involving psychotropic medications.
Serotonin Syndrome
Serotonin syndrome is a potentially fatal ADR that may induce altered mental status, neuromuscular irregularities, and autonomic hyperactivity. It occurs when an excess amount of serotonin accumulates in the body and overstimulates the central and peripheral serotonin receptors. The clinical presentation of serotonin syndrome varies, ranging from mild, nonspecific symptoms to a life-threatening condition. Symptoms usually present within the first 24 hours of the interaction and onset is generally rapid, sometimes occurring within minutes after ingestion of the offending agent. Milder cases of serotonin syndrome may be characterized by tremors, sweating, hypertension, and tachycardia, whereas life-threatening cases may present with delirium, muscle rigidity, convulsions, and hyperthermia.43 Management depends on the severity of the case, but the primary strategy is to remove the precipitating agent and provide supportive care.43
Numerous drugs have been implicated in the causation of serotonin syndrome. The drugs most commonly associated with it have serotonin-enhancing properties and include SSRIs, SNRIs, TCAs, monoamine oxidase inhibitors (MAOIs), buspirone, fentanyl, tramadol, St. John’s wort, lithium, ondansetron, and linezolid.15,43 The antidepressant class contains several drugs with the potential to cause serotonin syndrome when combined with other serotonergic drugs. In 2002, more than 26,000 incidences of serotonin syndrome linked to SSRI exposure were reported and resulted in more than 7,300 significant ADRs and 90 deaths.44 Additionally, several cases of serotonin syndrome have been reported for MAOIs used with TCAs or SSRIs, indicating that these combinations should be avoided.15 Drugs that inhibit the metabolism of specific antidepressants with serotonergic properties, especially CYP2D6 and CYP3A4 inhibitors, have a higher inherent potential to cause serotonin syndrome.43 The SNRIs venlafaxine and duloxetine are powerful serotonin reuptake inhibitors and therefore are key contributors to the occurrence of serotonin syndrome in patients taking other serotonergic drugs. Accordingly, the coadministration of SNRIs with SSRIs, TCAs, or MAOIs is contraindicated.5 Mirtazapine and trazodone are the antidepressants least likely to mediate PD interactions resulting in serotonin syndrome, but their use with other serotoninergic drugs is not without risk.
Hypotension and Fall Risk
PD interactions involving psychotropic medications that may result in hypotension can occur with treatment and are of particular concern in the elderly population because of the risk of falls and injury. This interaction is more likely to occur with drugs with pronounced antiadrenergic activity. Common agents implicated in this type of interaction are antipsychotics and BZDs.45
All antipsychotics carry a risk of orthostatic hypotension. In the CATIE trial, which compared the tolerability of first-generation antipsychotics (FGAs) and second-generation antipsychotics (SGAs), the highest incidence of hypotension occurred with the SGAs clozapine, quetiapine, and iloperidone.46,47 Although data on FGAs are limited, studies of chlorpromazine and thioridazine have found that these agents carry a high risk of hypotension.48
When they prescribe psychotropic medications, clinicians should consider concomitant drugs and diseases in order to reduce the risk of hypotension and resultant patient harm. Disease states associated with autonomic failure may predispose patients to hypotension; examples of these conditions include diabetic neuropathy, alcoholic neuropathy, and Parkinson’s disease.48
The risk of falls also increases with the use of various psychotropic medications. BZD use is associated with sedation, fatigue, memory problems, and slowing of psychomotor function. These effects are especially problematic in elderly patients. When BZDs are used in combination with other central nervous system (CNS) depressants (e.g., antihistamines, barbiturates, narcotics, alcohol, other psychotropics), the effects are compounded and pose a significant risk to patients. Age-dependent effects on drug action also influence patient response to BZDs. In elderly patients, slowed neuronal function and liver dysfunction confer increased risk of ADRs when these patients are also receiving BZD therapy. If BZDs are used in elderly patients, it is recommended that agents with a shorter half-life be used and combination therapy with other CNS depressants be avoided, if possible, to prevent toxicity.49
Hematologic Effects
Hematologic abnormalities are common ADRs of many psychotropic medications and may be enhanced when these agents are used in patients with predisposing factors to hematologic dysfunction (e.g., related to concomitant diseases and/or drug therapy).50 Although drug-induced hematologic effects are rare, certain agents reviewed in this lesson carry a clinically significant risk. See TABLE 2 for a list of hematologic effects associated with various psychotropic medications.
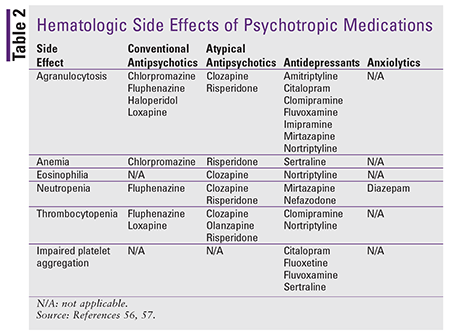
In the antipsychotic drug class, clozapine is the agent most significantly associated with agranulocytosis. It is recommended that providers closely monitor WBC counts and absolute neutrophil counts in all patients on clozapine therapy, especially during the first 3 months of therapy.51 It is prudent to consider discontinuing therapy if the patient becomes neutropenic.
A variety of hematologic effects can occur with antidepressants. Although newer classes of antidepressants carry a lower risk, older agents, such as TCAs, have been associated with agranulocytosis.52 Rare reports of neutropenia associated with mirtazapine are also found in the literature.53-55 SSRIs are linked to thrombocytopenia, and their effects on platelet aggregation may predispose patients to an increased risk of bleeding.56
Very little published data indicate a link between BZDs and hematologic abnormalities. Insufficient evidence exists that diazepam contributes to drug-induced neutropenia. The mechanism of this ADR is unknown and seems to be independent of daily dose or cumulative dose.57 Although these hematologic events are rare, the manufacturer of diazepam recommends routine CBC monitoring with long-term therapy.58
CONCLUSION
Drug interactions involving psychotropic medications may result in reduced therapeutic effects or an increased risk of ADRs, which influence patient outcomes. Most of these interactions involve PK pathways via CYP metabolism. In general, inhibition of CYP metabolism may increase drug concentrations and ADR risk, and induction may result in decreased therapeutic efficacy. Inhibition of intestinal Pgp may be linked to drug toxicity, whereas induction may influence clinical outcomes. PD interactions are often more complex and are identified when the clinician carefully assesses the net effect of combined therapies with either synergistic or antagonistic actions. Pharmacists should screen for these potential drug interactions and provide recommendations to appropriately address interactions that pose harm to patients. The best preventive strategies may start by identifying unnecessary medications.
REFERENCES
- Mental Health America. The state of mental health in America. www.mentalhealthamerica.net/issues/state-mental-health-america. Accessed August 21, 2017.
- Moore TJ, Mattison DR. Adult utilization of psychiatric drugs and differences by sex, age, and race. JAMA Intern Med. 2017;177:274275.
- CDC. CDC mental illness surveillance. www.cdc.gov/mentalhealthsurveillance/fact_sheet.html. Accessed August 21, 2017.
- McFarland HM. Identification and management of drug interactions. Medscape. www.medscape.org/viewarticle/418376. Accessed August 18, 2017.
- Ereshefsky L, Jhee S, Grothe D. Antidepressant drug-drug interaction profile update. Drugs R D. 2005;6:323-336.
- Johnson JA, Bootman JL. Drug-related morbidity and mortality and the economic impact of pharmaceutical care. Am J Health Syst Pharm. 1997;54:554-558.
- Strain JJ, Chiu NM, Sultana K, et al. Psychotropic drug versus psychotropic drug–update. Gen Hosp Psychiatry. 2004;26:87-105.
- Ereshefsky L. Drug-drug interactions with the use of psychotropic medications. Interview by Diane M. Sloan. CNS Spectr. 2009;14:1-8.
- Yap KY, Tay WL, Chui WK, Chan A. Clinically relevant drug interactions between anticancer drugs and psychotropic agents. Eur J Cancer Care (Engl). 2011;20:6-32.
- Guerra T. The top 200 drugs of 2017? Pharmacy Times. www. pharmacytimes.com/contributor/tony-guerra-pharmd/2017/03/the-top200-drugs-of-2017. Accessed August 10, 2017.
- Chadwick B, Waller DG, Edwards JG. Potentially hazardous drug interactions with psychotropics. Adv Psychiatr Treat. 2005;11:440449.
- Sikka R, Magauran B, Ulrich A, Shannon M. Bench to bedside: pharmacogenomics, adverse drug interactions, and the cytochrome p450 system. Acad Emerg Med. 2005;12:1227-1235.
- Gonzalez FJ, Coughtrie M, Tukey RH. Drug metabolism. In: Brunton LL, Chabner BA, Knollmann BC, eds. Goodman & Gilman’s The Pharmacological Basis of Therapeutics. 12th ed. New York, NY: McGraw-Hill. Accessed August 21, 2017.
- Tsunoda SM, Bednarczyk D, Okochi H. Drug transporters. In: Bertino JS Jr, Kashuba AD, Ma JD, et al, eds. Pharmacogenomics: An Introduction and Clinical Perspective. New York, NY: McGraw-Hill; 2013.
- Bleakley S. Antidepressant drug interactions: evidence and clinical significance. Prog Neurol Psychiatr. 2016;20:21-27.
- Spina E, Scordo MG. Clinically significant drug interactions with antidepressants in the elderly. Drugs Aging. 2002;19:299-320.
- Effexor XR (venlafaxine) package insert. Philadelphia, PA: Wyeth Pharmaceuticals, Inc; March 2017.
- Guzman F. Venlafaxine and desvenlafaxine: differences and similarities. https://psychopharmacologyinstitute.com/antidepressants/snris/ venlafaxine-desvenlafaxine-differences-similarities/. Accessed August 26, 2017.
- Spina E, Trifirò G, Caraci F. Clinically significant drug interactions with newer antidepressants. CNS Drugs. 2012;26:39-67.
- Preskorn S, Werder S. Detrimental antidepressant drug-drug interactions: are they clinically relevant? Neuropsychopharmacology. 2006;31:1605-1612.
- Gillman PK. Tricyclic antidepressant pharmacology and therapeutic drug interactions updated. Br J Pharmacol. 2007;151:737-748.
- Kennedy SH, McCann SM, Masellis M, et al. Combining bupropion SR with venlafaxine, paroxetine, or fluoxetine: a preliminary report on pharmacokinetic, therapeutic, and sexual dysfunction effects. J Clin Psychiatry. 2002;63:181-186.
- Owen JR, Nemeroff CB. New antidepressants and the cytochrome P450 system: focus on venlafaxine, nefazodone, and mirtazapine. Depress Anxiety. 1998;7(suppl 1):24-32.
- Wright CE, Lasher-Sisson TA, Steenwyk RC, Swanson CN. A pharmacokinetic evaluation of the combined administration of triazolam and fluoxetine. Pharmacotherapy. 1992;12:103-106.
- Moskowitz H, Burns M. The effects on performance of two antidepressants, alone and in combination with diazepam. Prog Neuropsychopharmacol Biol Psychiatry. 1988;12:783-792.
- Fleishaker JC, Hulst LK. A pharmacokinetic and pharmacodynamic evaluation of the combined administration of alprazolam and fluvoxamine. Eur J Clin Pharmacol. 1994;46:35-39.
- Kiang TK, Ensom MH, Chang TK. UDP-glucuronosyltransferases and clinical drug-drug interactions. Pharmacol Ther. 2005;106:97-132.
- Buspirone package insert. Princeton, NJ: Bristol-Myers Squibb Co; 2000.
- Wetzel H, Anghelescu I, Szegedi A, et al. Pharmacokinetic interactions of clozapine with selective serotonin reuptake inhibitors: differential effects of fluvoxamine and paroxetine in a prospective study. J Clin Psychopharmacol. 1998;18:2-9.
- Hiemke C, Weigmann H, Härtter S, et al. Elevated levels of clozapine in serum after addition of fluvoxamine. J Clin Psychopharmacol. 1994;14:279-281.
- Jerling M, Lindström L, Bondesson U, Bertilsson L. Fluvoxamine inhibition and carbamazepine induction of the metabolism of clozapine: evidence from a therapeutic drug monitoring service. Ther Drug Monit. 1994;16:368-374.
- Dequardo JR, Roberts M. Elevated clozapine levels after fluvoxamine initiation. Am J Psychiatry. 1996;153:840-841.
- DuMortier G, Lochu A, Colen de Melo P, et al. Elevated clozapine plasma concentrations after fluvoxamine initiation. Am J Psychiatry. 1996;153:738-739.
- Koponen HJ, Leinonen E, Lepola U. Fluvoxamine increases the serum clozapine levels significantly. Eur Neuropsychopharmacol. 1996;6:69-71.
- Szegedi A, Anghelescu I, Wiesner J, et al. Addition of low-dose of fluvoxamine to low-dose clozapine monotherapy in schizophrenia: drug monitoring and tolerability data from a prospective clinical trial. Pharmacopsychiatry. 1999;32:148-153.
- Fabrazzo M, La Pia S, Monteleone P, et al. Fluvoxamine increases plasma and urinary levels of clozapine and its major metabolites in a time- and dose-dependent manner. J Clin Psychopharmacol. 2000;20:708-710.
- Weigmann H, Gerek S, Zeisig A, et al. Fluvoxamine but not sertraline inhibits the metabolism of olanzapine: evidence from a therapeutic drug monitoring service. Ther Drug Monit. 2001;23:410-413.
- de Jong J, Hoogenboom B, van Troostwijk LD, de Haan L. Interaction of olanzapine with fluvoxamine. Psychopharmacology (Berl). 2001;155:219-220.
- Hiemke C, Peled A, Jabarin M, et al. Fluvoxamine augmentation of olanzapine in chronic schizophrenia: pharmacokinetic interactions and clinical effects. J Clin Psychopharmacol. 2002;22:502-506.
- Liu HC, Lin SK, Sung SM. Extrapyramidal side-effect due to drug combination of risperidone and donepezil. Psychiatry Clin Neurosci. 2002;56:479.
- Meyer JM, Loebel AD, Schweizer E. Lurasidone: a new drug in development for schizophrenia. Expert Opin Investig Drugs. 2009;18:1715-1726.
- Aripiprazole package insert. Princeton, NJ: Bristol-Myers Squibb Co; 2003.
- Boyer EW, Shannon M. The serotonin syndrome. New Engl J Med. 2005:352;1112-1120.
- Watson WA, Litovitz TL, Rodgers GC Jr, et al. 2002 annual report of the American Association of Poison Control Centers Toxic Exposure Surveillance System. Am J Emerg Med. 2003;21:353-421.
- de Jong MR, Van der Elst M, Hartholt KA. Drug-related falls in older patients: implicated drugs, consequences, and possible prevention strategies. Ther Adv Drug Saf. 2013;4:147-154.
- Stroup TS, Lieberman JA, McEvoy JP, et al. Results of phase 3 of the CATIE schizophrenia trial. Schizophr Res. 2009;107:1-12.
- Stroup TS, Lieberman JA, McEvoy JP, et al. Effectiveness of olanzapine, quetiapine, and risperidone in patients with chronic schizophrenia after discontinuing perphenazine: a CATIE study. Am J Psychiatry. 2007;164:415-427.
- Gugger JJ. Antipsychotic pharmacotherapy and orthostatic hypotension: identification and management. CNS Drugs. 2011;25:659-671.
- Díaz-Gutiérrez M, Martínez-Cengotitabengoa M, Sáez de Adana E, et al. Relationship between the use of benzodiazepines and falls in older adults: a systematic review. Maturitas. 2017;101:17-22.
- Stübner S, Grohmann R, Engle R, et al. Blood dyscrasias induced by psychotropic drugs. Pharmacopsychiatry. 2004;37(suppl 1):S70-S78.
- Clozaril (clozapine) package insert. East Hanover, NJ: Novartis Pharmaceutical Corp; 2014.
- Dodd S, Malhi GS, Tiller J, et al. A consensus statement for safety monitoring guidelines of treatments for major depressive disorder. Aust N Z J Psychiatry. 2011;45:712-725.
- Civalier KA, Krahn LE, Agrwal N. Repeated episodes of neutropenia triggered by mirtazapine. Psychosomatics. 2009;50:299-300.
- Ozcanli T, Unsalver B, Ozdemir S, Ozmen M. Sertraline- and mirtazapine-induced severe neutropenia. Am J Psychiatr. 2005;162:1386.
- Anghelescu I, Klawe C, Dahmen N. Venlafaxine in a patient with idiopathic leukopenia and mirtazapine-induced severe neutropenia. J Clin Psychiatry. 2002;63:838.
- McCloskey DJ, Postolache TT, Vittone BJ, et al. Selective serotonin reuptake inhibitors (SSRIs): measurement of effect on platelet function. Transl Res. 2008;151:168-172.
- Oyesanmi O, Kunkle E, Monti D, Field HL. Hematological side effects of psychotropics. Psychomatics. 1999;40:414-421.
- Diazepam package insert. Basel, Switzerland: Hoffman-La Roche; 2016.
- PL Detail-Document. Cytochrome P450 drug interactions. Pharmacist’s Letter/Prescriber’s Letter. May 2016.