Recently Approved Drugs for Health Issues Affecting Women
RELEASE DATE
September 1, 2022
EXPIRATION DATE
September 30, 2024
FACULTY
Justin J. Sherman, PharmD, MCS
Associate Professor of Pharmacy Practice
University of Mississippi School of Pharmacy
Jackson, Mississippi
FACULTY DISCLOSURE STATEMENTS
Dr. Sherman has no actual or potential conflicts of interest in relation to this activity.
Postgraduate Healthcare Education, LLC does not view the existence of relationships as an implication of bias or that the value of the material is decreased. The content of the activity was planned to be balanced, objective, and scientifically rigorous. Occasionally, authors may express opinions that represent their own viewpoint. Conclusions drawn by participants should be derived from objective analysis of scientific data.
ACCREDITATION STATEMENT
 Pharmacy
Pharmacy
Postgraduate Healthcare Education, LLC is accredited by the Accreditation Council for Pharmacy Education as a provider of continuing pharmacy education.
UAN: 0430-0000-22-089-H01-P
Credits: 2.0 hours (0.20 ceu)
Type of Activity: Knowledge
TARGET AUDIENCE
This accredited activity is targeted to pharmacists. Estimated time to complete this activity is 120 minutes.
Exam processing and other inquiries to:
CE Customer Service: (800) 825-4696 or cecustomerservice@powerpak.com
DISCLAIMER
Participants have an implied responsibility to use the newly acquired information to enhance patient outcomes and their own professional development. The information presented in this activity is not meant to serve as a guideline for patient management. Any procedures, medications, or other courses of diagnosis or treatment discussed or suggested in this activity should not be used by clinicians without evaluation of their patients’ conditions and possible contraindications or dangers in use, review of any applicable manufacturer’s product information, and comparison with recommendations of other authorities.
GOAL
To review selected drugs approved by the FDA over the past 2 years for the treatment of health issues affecting women.
OBJECTIVES
After completing this activity, the participant should be able to:
- Discuss the use of vibegron for overactive bladder and the use of ibrexafungerp for vulvovaginal candidiasis.
- Review the roles of eptinezumab-jjmr, atogepant, and rimegepant in migraine treatment.
- Discuss the drospirenone and estetrol combination oral contraceptive relative to other birth control agents.
- Describe new therapies for multiple sclerosis and for breast cancer.
ABSTRACT: Over the past 2 years, several new drugs have been approved and marketed for the management of disease states associated with women’s health. These include agents for overactive bladder, vulvovaginal candidiasis, migraine, breast cancer, and multiple sclerosis. Additionally, a new combined oral contraceptive has come to market. Several of these recently approved agents may provide better alternatives to current therapy, and some of them possess mechanisms of action that make them unique and interesting options. Pharmacists should educate themselves regarding the dosing and administration of these drugs; they should also determine whether a newer agent would be preferable to an established option in individual patients.
A multitude of drugs have been approved by the FDA over the past 2 years. Several of these drugs have novel mechanisms of action (MOAs) or possible enhancements over older agents with a slightly different MOA that have relevance for a number of women’s health issues.1,2 For example, three new agents may make an impact on the treatment of breast cancer. New drugs for vulvovaginal candidiasis and overactive bladder have been approved, as well as a new combined oral contraceptive. Additionally, three drugs for migraine and two for multiple sclerosis (MS) will be discussed because these diseases affect three times as many women as men. The purpose of this lesson is to review these recently approved drugs and discuss their possible role alongside existing treatment modalities.
Vibegron
Vibegron (Gemtesa; Urovant Sciences), a new beta-3 (B3) agonist, was approved by the FDA for treatment of overactive bladder on December 23, 2020.3 Vibegron is the first new oral medication (tablet) for overactive bladder to come to market in nearly a decade and the first to have no blood pressure warning in its label. It is also the second FDA-approved beta-3 agonist (mirabegron [Myrbetriq] is the first).3 Similar to mirabegron, vibegron has a relaxant effect on the bladder detrusor muscle.4
The recommended dosage of vibegron is 75 mg once daily. The pharmacist should counsel patients to swallow the tablet whole with water, without regard to food. If necessary, however, the tablet may be crushed and mixed with applesauce.4
Vibegron may increase the likelihood of urinary retention, especially if taken concomitantly with a muscarinic antagonist (e.g., oxybutynin, tolteridine).4 Vibegron must be discontinued upon any signs or symptoms of urinary retention. At least 2% of trial subjects experienced upper respiratory infection, nausea, diarrhea, and urinary tract infections. Headache was the most common adverse reaction (in 4%). Xerostomia may occur, although this is rare, and normal salivary flow resumes when vibegron is discontinued.4
Drug interactions include a possible increase in serum digoxin concentration. Serum digoxin concentrations should be monitored before vibegron therapy is initiated so that the digoxin can be titrated down, if needed. Vibegron is not approved for pediatric patients owing to a lack of safety and effectiveness data. Additionally, vibegron use is not recommended in patients with end-stage renal disease (ESRD) or severe hepatic impairment.4
The B3 agonists are alternatives to antimuscarinics.5 Vibegron may possess a slight advantage over mirabegron because, unlike the latter agent, its side-effect profile does not include hypertension or tachycardia. Although both vibegron and mirabegron alter digoxin concentrations, vibegron does not interact with CYP2D6 inhibitors. Vibegron is slightly more expensive than mirabegron. Antimuscarinics, still the mainstay of therapy, are inexpensive and effective but are limited by a comparatively worse adverse-event profile.5
All medications for overactive bladder are modestly effective, reducing incontinence episodes from three times per day to one or two times per day. The selected agent should be discontinued after 3 months if no benefit occurs. If monotherapy is not effective, combination treatment with antimuscarinics and B3 agonists may yield a slightly greater benefit.6 Pharmacists should also counsel patients regarding nonpharmacologic measures they can take to help relieve overactive bladder, such as scheduling urination, limiting caffeine intake, and avoiding fluids at bedtime.
Ibrexafungerp
Ibrexafungerp (Brexafemme; Scinexis) is the first new antifungal class (triterpenoid antifungal) approved for the treatment of vulvovaginal candidiasis (i.e., vaginal yeast infection) in more than 20 years.7 On June 1, 2021, the FDA approved the New Drug Application for this nonazole prescription medication for use in adult and postmenarchal female pediatric patients. The uniqueness of this new oral agent lies in its fungicidal activity against Candida species as opposed to simply growth inhibition.7
Prior to the approval of ibrexafungerp, fluconazole (Diflucan) was the only oral agent available for this indication. Ibrexafungerp inhibits glucan synthase (1,3-beta-d-glucan), an enzyme involved in the formation of an essential component of the fungal-cell wall. Glucan synthase is also the target of the fungicidal echinocandins (e.g., caspofungin). Ibrexafungerp is structurally distinct, however, and unique in its bioavailability because echinocandins are administered IV only.8
Ibrexafungerp is available as 150-mg tablets that may be taken with or without food. The recommended dosage for adults and postmenarchal female pediatric patients is two tablets twice daily for 1 day for a total treatment dosage of 600 mg. The prescriber should verify the patient’s pregnancy status prior to treatment, and effective contraception should be used during treatment. Based on animal studies, pregnancy is contraindicated during therapy because of the risk of fetal toxicity. Adverse reactions are mainly gastrointestinal: nausea, vomiting, dizziness, diarrhea, and abdominal pain.9
Ibrexafungerp exposure may be increased or decreased with concomitant use of strong CYP3A inhibitors (e.g., erythromycin, ketoconazole, clarithromycin, verapamil) or CYP3A inducers (e.g., phenytoin, carbamazepine, rifampin), respectively. With concomitant use of a strong CYP3A inhibitor, the ibrexafungerp dosage should be reduced to 150 mg twice daily for 1 day. The use of CYP3A inducers should be avoided during ibrexafungerp therapy.9
For lingering or difficult-to-treat vulvovaginal candidiasis, fluconazole has been dosed at one tablet twice weekly or one tablet weekly for 3 weeks, but more difficult infections have recently emerged. Ibrexafungerp may be useful when fluconazole is no longer as effective for a patient with a history of multiple infections. Ibrexafungerp fights more Candida strains, and symptom relief generally occurs within 10 days. However, at $475 for a four-tablet treatment, it may remain an alternative to fluconazole, which is less expensive. Regardless, as new multidrug-resistant Candida species begin to emerge, ibrexafungerp may come to be used more frequently based on its promise in treating these infections.8
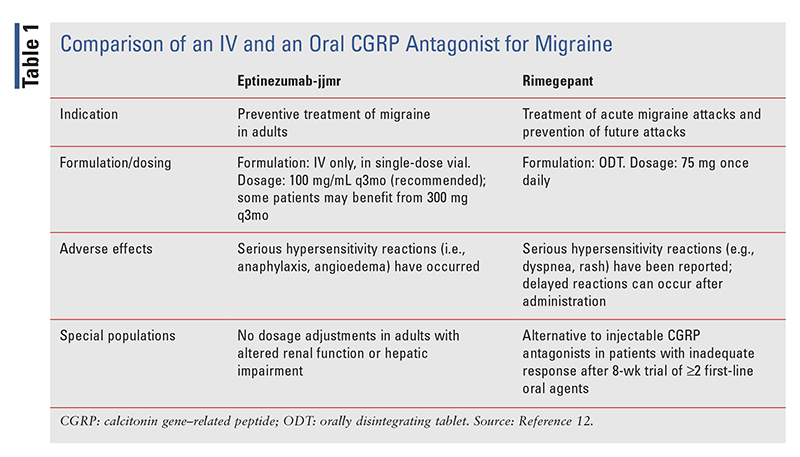
Eptinezumab-jjmr
Approved by the FDA on February 21, 2020, eptinezumab-jjmr (Vyepti; Lundbeck Seattle BioPharmaceuticals) is a calcitonin gene–related peptide (CGRP) antagonist indicated for preventive treatment of migraine in adults.10 CGRP antagonists have emerged over the past several years for migraine prophylaxis in injectable form. Others agents in this class include erenumab-aooe (Aimovig), fremanezumab-vfrm (Ajovy), and galcanezumab-gnlm (Emgality). CGRP, a potent vasodilator, is produced in peripheral and central nervous system (CNS) neurons. CGRP levels increase during an active migraine attack and then return to normal levels after the attack. This peptide is thought to play a role in the cardiovascular (CV) system as well.
Eptinezumab is administered IV once every 3 months in the prescriber’s office or other clinical setting.11 During administration, 100 mg/mL from a single-dose vial is infused after dilution with 100 mL of saline over 30 minutes. The vial should be gently inverted with diluted solution to mix; it should not be shaken. It is crucial not to administer this agent as an IV push or bolus injection. A regimen of 300 mg every 3 months may be beneficial for some patients. No dose adjustment is needed for altered renal function or hepatic impairment.11
Serious hypersensitivity reactions, such as anaphylaxis or angioedema, may occur.11 As a humanized monoclonal antibody (Mab), eptinezumab may cause placental transfer of human immunoglobulin G. A low exposure would be expected, and agents other than eptinezumab-jjmr would be a better choice for migraine prophylaxis in pregnant women. Nasopharyngitis is another adverse reaction.11 Although theoretical concerns exist for cardiac adverse effects, eptinezumab-jjmr does not need to be monitored for hypertension, unlike erenumab-aooe.12
Eptinezumab begins working to prevent migraine headache pain as quickly as the first day or week after the patient’s first infusion.12 Many patients experience efficacy before 30 days of use. Compared with placebo, CGRP antagonists prevent one to two more episodic migraines per month, and they reduce migraine days per month by at least 50% in almost 25% of patients. The main disadvantage of this agent is that it must be administered in a clinical setting, whereas many other preventives can be self-administered.12
The cost of eptinezumab is high at $18,000 per year, versus a cost of approximately $7,200 per year for other injectable preventives in this CGRP class.12 Because of its expense, patients may first need to fail historically used agents for migraine prophylaxis (e.g., propranolol, topiramate) before insurance companies will pay for CGRP antagonists.12
Atogepant
On September 28, 2021, the FDA approved atogepant (Qulipta; AbbVie) for the preventive treatment of episodic migraine in adults.13 This agent is the second oral CGRP antagonist approved for migraine prophylaxis. (Rimegepant, which is discussed below, was the first.)13
Atogepant is dosed at one tablet per day, with three different strengths available: 10 mg, 30 mg, and 60 mg.14 It may be taken with or without food and is absorbed with peak plasma concentrations at approximately 1 to 2 hours. Atogepant did not have a significant food effect when administered with a high-fat meal; the AUC and maximum concentration of drug were reduced by approximately 18% and 22%, respectively.
Adverse reactions of atogepant include nausea, constipation, and fatigue.14 In placebo-controlled clinical trials, transaminase elevations exceeding three times the upper limit of normal were temporally associated with atogepant treatment, but the elevation resolved within 8 weeks of discontinuation, and there were no cases of severe liver injury or jaundice. Based on the occurrence of transaminase elevations in these trials, atogepant use should be avoided in patients with severe hepatic impairment. Dosing should be limited to 10 mg daily in patients with ESRD or severe renal disease.14
Atogepant interacts with CYP3A4 inhibitors and inducers. Recommended dosing is 10 mg daily when it is given concomitantly with CYP3A4 inhibitors, and 30 mg or 60 mg daily when given with inducers.14 With the use of organic anion transport protein inhibitors such as cyclosporin, atogepant dosing should be limited to 10 mg or 30 mg once daily. Based on animal data, the use of this drug is not recommended in pregnant women.14
Rimegepant
Rimegepant (Nurtec ODT [orally disintegrating tablet]; Biohaven Pharmaceuticals) is the only migraine medication that is approved for both the treatment of acute migraine attacks and the prevention of future migraine attacks.15 The FDA approved rimegepant for acute treatment of migraine with or without aura on February 27, 2020, and for prevention of episodic migraine on May 27, 2021. It is the first CGRP antagonist available as a fast-acting ODT.15 Ubrogepant (Ubrelvy) is comparable for acute treatment, and atogepant is comparable for migraine prophylaxis.
For acute treatment, the recommended dosage is 75 mg orally as needed; for prophylaxis, it is 75 mg every other day.16 The maximum dosage in a 24-hour period is 75 mg. Patients should be counseled not to take more than 18 doses per month because the safety of exceeding that amount has not been established. The tablet should be immediately placed on or under the tongue, where it dissolves and may be swallowed without additional liquid; it may be administered without regard to food.16
This agent is contraindicated in patients with a history of hypersensitivity reaction to rimegepant or any of its components.16 Hypersensitivity reactions such as dyspnea and rash have been reported, and delayed reactions have sometimes occurred after administration. Nausea and abdominal pain are the most commonly reported adverse effects. Strong and moderate CYP3A4 inhibitors and CYP3A4 inducers should be avoided because the dose cannot be adjusted. P-glycoprotein or breast cancer resistance protein inhibitors (e.g., protease inhibitors, tyrosine kinase inhibitors) should also be avoided. Patients with severe hepatic disease or ESRD should not receive rimegepant. The risks of using this drug during pregnancy or while breastfeeding are unknown.16
As an oral medication, rimegepant has a key advantage over preventive CGRP antagonists that are administered IV.12 As noted above, epitnezumab-jjmr is one of these IV agents. TABLE 1 presents a comparison of epitnezumab-jjmr and rimegepant.12
Drospirenone and Estetrol
Drospirenone 3 mg and estetrol (DRSP and E4) 14.2 mg (Nextstellis; Mayne Pharma) is a new combined oral contraceptive (COC).17 Approved by the FDA on April 16, 2021, this product is the first and only contraceptive pill that contains E4; most short-acting combination contraceptives contain progesterone and ethinyl estradiol. This COC may be less effective in females with a BMI of 30 kg/m2 or higher, as its efficacy is inversely proportional to increased BMI.
The synthetic estrogen E4 is hypothesized to bind widely to all estrogen receptors in the body. Although E4 is naturally produced in the body, the E4 component in this medication is made from a plant source. In trials, this COC met efficacy end points for prevention of pregnancy, and it also met secondary end points for bleeding, safety, and tolerability.18
The usual dosage of DRSP and E4 is 24 active (pink) tablets once daily followed by four inactive (white) tablets once daily.19 For patients not currently using a hormonal contraceptive, the first active tablet should be taken on the first day of the menstrual cycle (i.e., day 1 start). If administration is not initiated on the first day of menses, an additional method of nonhormonal contraception must be used for the first 7 days of consecutive administration. If one active tablet is missed, the patient should take the missed dose as soon as possible. The remaining doses should be taken at the usual time, even if that means two doses on the same day. In postpartum women, administration should not be initiated earlier than 4 weeks following delivery.19
As with other COCs, this agent has a black box warning (BBW) contraindicating its use in females older than age 35 years who smoke because of the increased risk of CV events. Other contraindications include patients with high risk of arterial or venous thrombotic disease; a history of or current hormonally sensitive malignancy, including breast cancer; hepatic adenoma; coadministration with hepatitis C drug combinations; abnormal uterine bleeding; renal impairment; and adrenal insufficiency.19
Bleeding irregularities may occur during the first 4 months of therapy, and amenorrhea or oligomenorrhea may occur after discontinuation.19 The DRSP and E4 combination has been associated with a slightly increased risk of cervical cancer. This agent has antimineralcorticoid activity that may lead to hyperkalemia. Patients with hypertriglyceridemia or a family history of it may be at increased risk for pancreatitis while taking this medication. The DRSP and E4 pill should be discontinued if thrombotic events occur.19
Warnings and precautions include thrombotic disorders or other vascular problems, and this COC should be stopped if a thrombotic event occurs.19 All CV risk factors should be considered before initiating this agent in any patient, particularly if multiple risk factors exist. Because of the increased risk of hyperkalemia, patients taking medications that may increase the potassium concentration (e.g., ACE inhibitors, spironolactone) should be monitored, preferably at the start of the next cycle of the COC. The DRSP and E4 pill should be discontinued if the patient is diagnosed with breast cancer or develops significant liver disease, hypertriglyceridemia, or cholestasis. Patients should be monitored for significant hypertension, migraine, or bleeding irregularities.19
Adverse reactions include bleeding irregularities, headache, dysmenorrhea, increased weight, mood disturbances, breast symptoms, acne, and decreased libido.19 Concomitant use of CYP3A inducers may result in contraceptive failure and breakthrough bleeding. The patient should avoid concomitant use or should use an alternative or back-up contraceptive method during coadministration and for up to 28 days after stopping the CYP3A inducer. DRSP and E4 must be discontinued if pregnancy occurs, and this COC can reduce milk production in postpartum women.19
Because this DRSP and E4 combination is the first oral contraceptive pill in decades to contain a new estrogen, pharmacists should be aware of certain considerations. In contrast to ethinyl estradiol, which is dosed in micrograms, estetrol is dosed is in milligrams; this makes it more difficult to compare DRSP and E4 with other COCs.18 This agent seems as effective as other COCs, but it is theorized that E4 will have less impact on certain tissues or metabolic changes. At this time, insufficient evidence exists on whether it decreases the risks of breast cancer and blood clots compared with other combination COCs. (These risks were not primary end points in studies.) Finally, the DRSP and E4 combination costs $190 per month, compared with a cost of less than $10 per month for some generics.18
Ponesimod
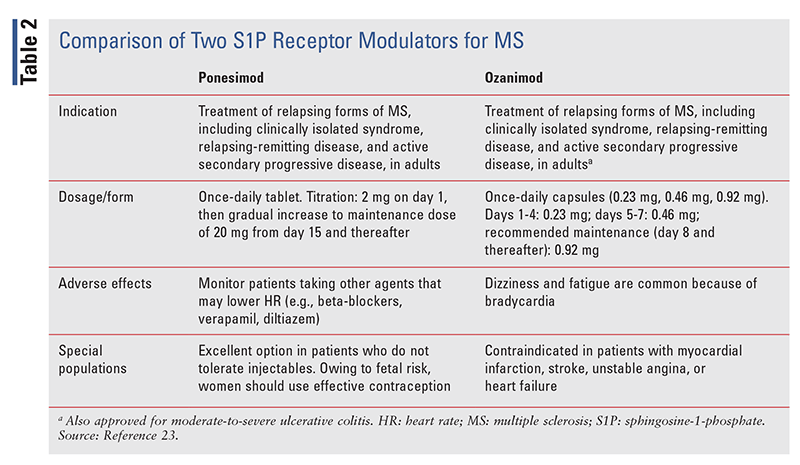
Ponesimod (Ponvory; Janssen Pharmaceuticals) is a sphingosine-1-phosphate (S1P) receptor modulator indicated for the treatment of relapsing forms of MS in adults.20,21 The FDA approved this agent on March 19, 2021, for clinically isolated syndrome, relapsing-remitting disease, and active secondary progressive disease.20,21 Comparable medications include siponimod (Mayzent), fingolimod (Gilenya), and ozanimod (Zeposia).
Although ponesimod’s MOA in MS is unknown, it may involve the reduction of lymphocyte migration into the CNS (i.e., suppressing migration of lymphocytes from lymph nodes to the location of inflammation). This S1P receptor 1 modulator binds with high affinity to S1P receptor 1. Ponesimod blocks the capacity of lymphocytes to exit the lymph nodes, reducing the number of lymphocytes in peripheral blood.21
Ponesimod is available as a titration starter pack of tablets ranging from 2 mg at day 1 up to 10 mg at days 12 through 14, and as 20-mg tablets for maintenance therapy starting at day 15. Dosing is once daily, every day. If more than four consecutive doses are missed, the patient should reinitiate treatment starting at day 1 of the initial titration regimen. If fewer than three consecutive doses are missed, the patient should resume with the first missed titration dose, then resume the titration schedule at that dose and titration day.22
Initiation of ponesimod has been associated with transient atrioventricular (AV) conduction delays, including first-degree AV block.22 First-dose monitoring for 4 hours at the prescriber’s office is recommended, with pulse and blood-pressure monitoring every hour. ECG monitoring should be conducted prior to dosing and after the first dose; gradual titration reduces cardiac effects. Contraindications include 2nd-degree or 3rd-degree AV block, sick sinus syndrome, and sinoatrial block within 6 months of therapy initiation. Also, ponesimod is contraindicated if the patient had a myocardial infarction, unstable angina, stroke, decompensated heart failure with hospitalization, transient ischemic attack, or class III/IV heart failure within 6 months of therapy initiation. Patients taking concomitant medications that may decrease heart rate, such as beta-blockers, verapamil, and diltiazem, should be monitored.22
Warnings and precautions include infections (obtain pertinent laboratory values around treatment), liver injury (obtain liver-function tests prior to starting doses), and fetal risk (women should use effective contraception during and 1 week after stopping ponesimod). Bradyarrhythmia, increased blood pressure, cutaneous malignancies, and macular edema may also occur. An ophthalmologic examination should be conducted prior to initiation. Given the possibility of decreased pulmonary function, spirometry may be performed prior to therapy.22
The most common adverse reactions (in >10% of patients) are upper respiratory tract infections, elevation of hepatic transaminase, and hypertension.22 Live-attenuated vaccines should be avoided during treatment and for 7 to 14 days after therapy discontinuation. Owing to the increased infection risk, it is necessary to obtain a CBC before treatment initiation. Coadministration of ponesimod with strong CYP3A4 inducers should be avoided, and ponesimod is not recommended in patients with moderate or severe hepatic impairment.22
Ponesimod is an excellent option for patients who do not tolerate injectables. However, the monthly cost of this specialty medication is quite high. Although ponesimod lowers the number of WBCs in the blood, it also increases the risk of infection. Lymphocyte counts return to normal 1 week after stopping treatment, which is faster than in-class medications.23
Ozanimod
The FDA approved ozanimod (Zeposia; Celgene) on March 25, 2020, for the treatment of relapsing forms of MS in adults, including clinically isolated syndrome, relapsing-remitting disease, and active secondary progressive disease.24 (It was subsequently also approved for the treatment of moderate-to-severe ulcerative colitis.)24
Ozanimod is an S1P receptor modulator that binds with high affinity to S1P receptors 1 and 5. It blocks the capacity of lymphocytes to exit the lymph nodes, reducing the lymphocytes in peripheral blood. Ozanimod has minimal or no activity on S1P2, S1P3, and S1P4 receptors; the clinical relevance of its binding with high affinity to S1P5 receptors is unknown. The MOA may involve the reduction of lymphocyte migration into the CNS and intestine.24
Ozanimod, a capsule taken once daily, is available in three strengths: 0.23 mg, 0.46 mg, and 0.92 mg. For the first 4 days of titration the dosage is 0.23 mg, followed by a dosage of 0.46 mg for the next 3 days.24 The recommended dosage for maintenance therapy (i.e., day 7 and thereafter) is 0.92 mg. If a dose is missed during the first 2 weeks of treatment, the titration regimen must be restarted; if a dose is missed after the first 2 weeks, maintenance therapy may be continued without interruption.24
Selective serotonin reuptake inhibitors and serotoninnorepinephrine reuptake inhibitors may interact with ozanimod; therefore their coadministration with ozanimod is not recommended.24 Contraindications for ozanimod are the same as those noted above for ponesimod, with additional contraindications including severe, untreated sleep apnea and concomitant use of a monoamine oxidase inhibitor. Other warnings and precautions and use in special populations are identical to those listed above for ponesimod. Titration of 7 days is crucial in order to reduce bradycardia. Assessments that should be performed prior to ozanimod initiation include CBC, ECG, liver-function testing, and an ophthalmic examination (in patients with macular edema). In terms of adverse reactions, trials of ozanimod for MS showed that at least 4% of subjects experienced upper respiratory infection, increased hepatic enzymes, orthostasis, urinary tract infection, back pain, or hypertension.24
TABLE 2 provides a comparison of ozanimod and ponesimod for the treatment of MS.23 Ozanimod is somewhat less expensive, at $7,000 per month versus $8,000 per month for ponesimod. The titration period for ozanimod is shorter, being 7 days compared with 2 weeks for ponesimod.23
Tucatinib
On April 17, 2020, tucatinib (Turkysa; SeaGenetics) received accelerated approval from the FDA. This third-generation human epidermal growth factor 2 (HER2) tyrosine kinase inhibitor (TKI) is used in combination with trastuzumab and capecitabine for the treatment of advanced unresectable or metastatic HER2-positive breast cancer, including patients with brain metastasis.25 Patients with brain metastasis should have previously received one or more anti-HER2 regimens in a metastatic setting.25
Tucatinib is known for having a manageable toxicity profile.26 This drug crosses into the cytoplasm and binds to the adenosine triphosphate–binding domain on the intracellular portion of the HER2 receptor, inhibiting phosphorylation and preventing activation of downstream signaling pathways. With trastuzumab, resistance can occur over time because of a conformational change occurring on the external domain of the HER2 receptor that prevents binding to trastuzumab. Tucatinib circumvents this issue by binding directly to the intracellular portion.26
Tucatinib is available in tablets of 50 mg and 150 mg.25 The recommended dosage is 300 mg twice daily, taken with or without food. In patients with severe hepatic impairment, the recommended dosage is 200 mg twice daily.25
Warnings and precautions surrounding tucatinib include severe diarrhea, hepatotoxicity, and embryo-fetal toxicity.25 Severe diarrhea could result in dehydration and death. It is important to monitor hepatic enzymes, including bilirubin, prior to administration and every 3 weeks during treatment. Embryo-fetal toxicity could reduce fetal weight and cause abnormalities. It is important for the patient to use contraceptives during treatment and the first week after treatment discontinuation.25 The dosing of tucatinib should be reduced, temporarily interrupted, or discontinued based on the severity of these problems.
A major adverse effect of tucatinib is palmar-plantar erythrodysesthesia (i.e., redness, swelling, and blistering on the palms and soles of the feet), a condition caused by some chemotherapeutic agents. Other adverse effects include diarrhea, nausea, fatigue, vomiting, stomatitis, decreased appetite, abdominal pain, headache, anemia, and rash.25
Since TKIs can cross into the cytoplasm, any conformational change in the extracellular domain is circumvented; also, TKIs cross the blood-brain barrier to target brain metastasis.26 Both of these represent notable advantages of tucatinib over Mabs. One disadvantage of tucatinib is that it interacts with CYP3A substrates such as simvastatin and lovastatin, so concomitant use should be avoided. Some CYP3A inducers, such as carbamazepine, rifampin, and phenytoin, also should be avoided in combination with tucatinib. Patients are also advised not to breastfeed during tucatinib therapy.26
Sacituzumab Govitecan-hziy
Sacituzumab govitecan-hziy (Trodelvy; Gilead Sciences), a trophoblast cell-surface antigen 2 (trop-2)–directed antibody and topoisomerase inhibitor conjugate, received FDA approval on April 7, 2021.27 One of this drug’s two indications is for unresectable locally advanced or metastatic triple-negative (estrogen hormone receptor–, HER2 receptor–, and progesterone hormone receptor–negative) breast cancer patients who have received two or more prior systemic therapies, at least one of them for metastatic disease. The second indication (approved under accelerated approval based on tumor response rate and duration of response) is for patients with locally advanced or metastatic urothelial cancer who have previously received a platinum-containing chemotherapy agent and either programmed death receptor-1 or programmed death-ligand 1 inhibitor.27
Trop-2 receptors are frequently expressed in multiple types of epithelial tumors. High expression is associated with poor survival and relapse.28 Sacituzumab consists of two connected parts: an antibody that attaches to a trop-2 on the surface of the breast cancer cells and a chemotherapy drug that kills cancerous cells.28
Sacituzumab is available as 180 mg lyophilized powder in single-dose vials.27 The recommended dosage is 10 mg/kg once weekly on days 1 and 8 of a continuous 21-day treatment cycle until disease progression or unacceptable toxicity occurs. This agent is for IV infusion only; therefore, it should not be administered as an IV push or bolus. Premedication is recommended for prevention of infusion reactions and chemotherapy-induced nausea and vomiting. The patient should be monitored during the infusion and for at least 30 minutes after its completion. Treatment interruption and/or dose reduction may be necessary to manage adverse reactions.27
This agent has BBWs for severe or life-threatening neutropenia and severe diarrhea.27 Treatment should be stopped if the absolute neutrophil count is less than 1,500 mm3. For severe diarrhea, fluids and electrolytes should be administered to lessen the chance of dehydration. If the patient had previous hypersensitivity and infusion-related reactions, antihistamines or corticosteroids may be given. Patients who are homozygous for the UGT1A1 allele have an increased risk of neutropenia. Sacituzumab can cause embryo-fetal toxicity, and it is recommended to avoid breastfeeding.27
Margetuximab-cmkb
Margetuximab-cmkb (Margenza; MacroGenics), a HER2/neu receptor antagonist, was approved by the FDA on December 16, 2020. This agent is indicated in combination with chemotherapy for treatment of metastatic HER-positive breast cancer in adults who received two or more prior anti-HER2 regimens, at least one of which for metastatic disease.29
Margetuximab is a novel Fc-engineered anti-HER2 Mab derivative of trastuzumb.30 Both margetuximab and trastuzumb bind to the same epitope of the HER2 receptor. Because margetuximab has a higher affinity to CD16A, which is a stimulatory receptor, it increases antibody-dependent cellular cytotoxicity.30
The recommended dosage of margetuximab is 15 mg/ kg administered as an IV infusion every 3 weeks (21-day cycle) until disease progression or unacceptable toxicity occurs.29 The initial dose should be administered over 120 minutes; all subsequent doses should be administered over a minimum of 30 minutes. The infusion should be administered immediately after completion of chemotherapy.29
Infusion-related reactions can occur with margetuximab use, and the labeling contains BBWs for left ventricular dysfunction and embryo-fetal toxicity.29 Because this drug may reduce left ventricular ejection fraction, cardiac function should be evaluated prior to and during treatment, and it should be discontinued if the decrease is clinically significant. Adverse reactions include fatigue, nausea, diarrhea, and palmar-plantar erythrodysesthesia. Contraception is necessary because of the possibility of embryo-fetal toxicity.29
Conclusion
A variety of recently approved drugs offer more options for disease states associated with women’s health; additionally, a new COC has come to market. Several of these agents may provide better alternatives to current therapy, and some of them possess MOAs that make them unique and interesting options. Pharmacists should educate themselves regarding the dosing and administration of these drugs; they also should determine whether a newer agent would be preferable to an established option in individual patients.
REFERENCES
- FDA Center for Drug Evaluation and Research. Advancing Health Through Innovation: New Drug Therapy Approvals 2021. Silver Spring, MD: FDA; 2022.
- FDA Center for Drug Evaluation and Research. Advancing Health Through Innovation: New Drug Therapy Approvals 2020. Silver Spring, MD: FDA; 2021.
- Urovant Sciences. Urovant Sciences announces U.S. commercial launch of GEMTESA® (vibegron) 75 mg tablets for patients with overactive bladder. https://media.urovant.com/news-releases/news-release-details/ urovant-sciences-announces-us-commercial-launch-gemtesar. Accessed June 30, 2022.
- Gemtesa (vibegron) package insert. Irvine, CA: Urovant Sciences, Inc; December 2020.
- Krhut J, Skugarevska B, Mika D, et al. Clinical utility of beta3-adrenoreceptor agonists for the treatment of overactive bladder: a review of the evidence and current recommendations. Res Rep Urol. 2022;14:167-175.
- Emphasize nondrug measures for overactive bladder…not meds. Pharm Lett. March 2021.
- Scynexis, Inc. SCYNEXIS announces U.S. availability of Brexafemme® (ibrexafungerp tablets), the first new antifungal class of therapy approved by the U.S. FDA for vaginal yeast infections in more than 20 years. www.scynexis.com/news-media/press-releases/detail/252/scynexisannounces-u-s-availability-of-brexafemme. Accessed June 26, 2022.
- Colombo RE, Vazquez JA. An evaluation of ibrexafungerp for the treatment of invasive candidiasis: the evidence to date. Expert Opin Pharmacother. 2021;22(7):797-807.
- Brexafemme (ibrexafungerp) package insert. Jersey City, NJ: Scynexis, Inc; June 2022.
- American Headache Society. Breaking news: eptinezumab-jjmr (Vyepti™) approved by FDA. https://americanheadachesociety.org/news/ eptinezumab-vyepti-approved-by-fda/. Accessed June 27, 2022.
- Vyepti (eptinezumab-jjmr) package insert. Bothell, WA; Lundbeck Seattle BioPharmaceuticals, Inc; April 2022.
- Drugs to prevent migraine in adults. Pharm Lett. November 2020.
- Optum. New options for preventive treatment of episodic migraine—Qulipta™ (atogepant) and Nurtec ODT® (rimegepant). https://workcompauto.optum.com/content/owca/owca/en/insights/blog/ clinical-connection-blog/2021/New-options-for-preventive-treatment-ofepisodic-migraine.html. Accessed June 28, 2022.
- Qulipta (atogepant) package insert. North Chicago, IL: AbbVie, Inc; October 2021.
- Brooks M. FDA expands rimegepant indication to include migraine prevention. www.medscape.com/viewarticle/952073. Medscape Medical News. Accessed June 29, 2022.
- Nurtec ODT (rimegepant) package insert. New Haven, CT: Biohaven Pharmaceuticals, Inc; April 2022.
- Carr L. FDA approves new oral contraceptive Nextstellis. Contemporary OB/GYN. www.contemporaryobgyn.net/view/fda-approves-new-oralcontraceptive-nextstellis. Accessed June 24, 2022.
- Expect questions about Nextstellis for contraception. Pharm Lett. July 2021.
- Nextstellis (drospirenone and estetrol) package insert. Greenville, NC: Mayne Pharma; August 2022.
- Pharmaceutical Technology. US FDA approves Janssen’s PONVORY for relapsing multiple sclerosis. www.pharmaceutical-technology.com/ news/fda-approves-janssen-ponvory/. Accessed June 23, 2022.
- Subei AM, Cohen JA. Sphingosine 1-phosphate receptor modulators in multiple sclerosis. CNS Drugs. 2015;29(7):565-575.
- Ponvory (ponesimod) package insert. Titusville, NJ: Janssen Pharmaceuticals, Inc; March 2021.
- Multiple sclerosis treatments. Pharm Lett. May 2020.
- Zeposia (ozanimod) package insert. Summit, NJ: Celgene Corp; April 2022.
- Tukysa (tucatinib) package insert. Bothell, WA: SeaGen Inc; February 2022.
- Urlich L, Okines AF. Treating advanced unresectable or metastatic HER2-positive breast cancer: a spotlight on tucatinib. Breast Cancer (Dove Med Press). 2021;13:361-381.
- Trodelvy (sacituzumab govitecan-hziy) package insert. Foster City, CA: Gilead Sciences, Inc; June 2022.
- Seligson JM, Patron AM, Berger MJ, et al. Sacituzumab govitecanhziy: an antibody-drug conjugate for the treatment of refractory, metastatic, triple-negative breast cancer. Ann Pharmacother. 2021;55(7):921931.
- Margenza (margetuximab-cmkb) package insert. Rockville, MD: MacroGenics, Inc; December 2020.
- Schlam I, Nunes R, Lynce F. Profile of margetuximab: evidence to date in the targeted treatment of metastatic HER2-positive breast cancer. Onco Targets Ther. 2022;15:471-478.