
Reviewing the Regulatory Landscape
RELEASE DATE:
October 1, 2017
EXPIRATION DATE:
October 31, 2019
FACULTY:
Gerald Gianutsos, PhD, JD
Emeritus Associate Professor
University of Connecticut School of Pharmacy
Storrs, Connecticut
FACULTY DISCLOSURE STATEMENTS:
Dr. Gianutsos has no actual or potential conflict of interest in relation to this activity.
Postgraduate Healthcare Education, LLC does not view the existence of relationships as an implication of bias or that the value of the material is decreased. The content of the activity was planned to be balanced, objective, and scientifically rigorous. Occasionally, authors may express opinions that represent their own viewpoint. Conclusions drawn by participants should be derived from objective analysis of scientific data.
ACCREDITATION STATEMENT:
Pharmacy
 Postgraduate Healthcare Education, LLC is accredited by the Accreditation Council for Pharmacy Education as a provider of continuing pharmacy education.
Postgraduate Healthcare Education, LLC is accredited by the Accreditation Council for Pharmacy Education as a provider of continuing pharmacy education.
UAN: 0430-0000-17-071-H03-P
Credits: 2.0 hours (0.20 ceu)
Type of Activity: Knowledge
TARGET AUDIENCE:
This accredited activity is targeted to pharmacists. Estimated time to complete this activity is 120 minutes.
Exam processing and other inquiries to:
CE Customer Service: (800) 825-4696 or cecustomerservice@jobson.com
DISCLAIMER:
Participants have an implied responsibility to use the newly acquired information to enhance patient outcomes and their own professional development. The information presented in this activity is not meant to serve as a guideline for patient management. Any procedures, medications, or other courses of diagnosis or treatment discussed or suggested in this activity should not be used by clinicians without evaluation of their patients' conditions and possible contraindications or dangers in use, review of any applicable manufacturer's product information, and comparison with recommendations of other authorities.
GOAL:
To improve the pharmacist's understanding of the process of approval of different classes of products available in the pharmacy and their regulatory oversight.
OBJECTIVES:
After completing this activity, the participant should be able to:
- Explain what is meant by the term drug.
- Describe the approval process for prescription, OTC, and homeopathic drugs.
- Differentiate a drug and a dietary supplement.
- Explain recent regulatory concerns about homeopathic products.
- Describe the role of the pharmacist in helping consumers understand the differences in FDA oversight among different drug products.
ABSTRACT: The FDA is responsible for the oversight of prescription, OTC, homeopathic, and dietary supplement products. However, all these products are regulated differently. Prescription drugs require rigorous premarket testing, providing proof of safety and efficacy, and must receive affirmative FDA approval. OTC drugs and homeopathic drugs have a less onerous burden of complying with monographs (standards) published by the FDA and the Homeopathic Pharmacopeia, respectively. Supplements do not require premarket FDA approval. However, recent regulatory actions by the FDA and the FTC may impact supplements and homeopathic drugs. Pharmacists are uniquely qualified to help consumers understand the implications of the approval process.
The FDA is responsible for regulating a wide range of consumer products. These include human and veterinary drugs, both prescription and OTC; foods (except for some meat, poultry, and egg products, which are regulated by the U.S. Department of Agriculture); animal food and feed; vaccines and other biological products, including blood and human tissue; dietary supplements; medical devices intended for human use; radiation-emitting electronic products, such as x-ray machines, microwave ovens, CD ROMs, and laser pointers; cosmetics (shampoos, face creams, and makeup, but not if it meets the regulatory definition of a soap, which is regulated by the Consumer Products Safety Commission); and color additives.1 Since 2009, the FDA has also regulated cigarettes and smokeless and roll-your-own tobacco. In 2016, its authority was expanded to all tobacco products, including cigars, e-cigarettes, hookahs, and pipe tobacco.2 In all, sales of the products that fall under the FDA’s authority account for one-quarter of the total consumer spending in the United States.3
These diverse products coming under the FDA’s jurisdiction are regulated differently. Pharmacists are generally aware of the requirement for rigorous testing, including extensive clinical trials, and review before approval for marketing a prescription drug is granted by the FDA. However, they may be less familiar with the oversight of other classes of medicinal substances, which are subject to different regulatory requirements and which, overall, are subject to less stringent requirements than those for prescription products. Consumers and other healthcare practitioners are largely unaware of these differences and the degree of oversight provided by the FDA, and they may look to the pharmacist to provide guidance. This article will review the regulation of prescription, OTC, and homeopathic drugs and dietary supplements, highlighting some recently enacted and proposed regulatory changes.
What Are Drugs?
One of the first benchmarks necessary to distinguish the regulation of different kinds of pharmacologically active available products is to appreciate what is meant by the term drug. An important criterion under the Federal Food, Drug, and Cosmetic Act (FDCA) and FDA regulations is the intended use of a product. Drugs are defined with reference to their intended use: Drugs are “articles intended for use in the diagnosis, cure, mitigation, treatment, or prevention of disease” (also known as the therapeutic or disease claim) and “articles (other than food) intended to affect the structure or any function of the body of man or other animals,” also known as the structure function claim.4 The FDA will look at relevant documentation from the manufacturer, including the labeling for a product, websites, promotional pamphlets, and other marketing material. If any labeling claims a therapeutic benefit, the FDA will regulate the product as a drug. Vitamins and certain other alternative products that do not make a therapeutic claim are not regulated as drugs (see below).
Prescription and OTC Drugs
Drugs are further classified as either requiring a prescription or being available OTC. (Controlled substances and the concept of behind-the-counter drugs are beyond the scope of this article.) Prescription and OTC drugs must meet different criteria before the FDA will approve them for marketing. The FDA was granted the authority to distinguish between Rx and OTC drugs with passage of the Durham-Humphrey Amendment to the FDCA in 1951.5 Prior to this amendment, there was no formal distinction between Rx and OTC drugs, and the same drug was often marketed as both an OTC and an Rx drug. The amendment established a legal framework to differentiate prescription and nonprescription drugs and authorized the FDA to make the distinction.5 The FDA makes the determination whether a drug will be marketed as Rx-only or OTC at the time that the manufacturer seeks approval to market the drug by filing a New Drug Application (NDA).
Prior to 1951, the manufacturer could decide whether a drug was going to be marketed as Rx or OTC. The FDA is given considerable latitude in making its decision. The Durham-Humphrey Amendment also prohibited a drug from being marketed as both an OTC and an Rx drug at the same dosage and indication, although there can be both an OTC and an Rx version of a drug if they have different dosages or indications (e.g., Nexium). In effect, the differentiation between drug classes is based upon the FDA’s determination of the potential toxicity of the drug and the degree of supervision needed to safely use the drug, taking into consideration both the type of drug and the risks of self-diagnosis by the patient. The FDA defines OTC drugs as “drugs that are safe and effective for use by the general public without seeking treatment by a health professional.”6
Pharmacists should recall that prescription drugs must undergo rigorous preclinical and clinical testing to provide evidence to the FDA of their efficacy and safety before approval is granted. This will not be reviewed here except for a reminder that this practice requires many years of clinical trials; the interested reader can consult other resources.4,7 (Pharmacists should also be aware that some drugs marketed before the enactment of the FDCA in 1938 were grandfathered and have never been approved by the FDA. See Reference 8.)
OTC Drugs
The FDA estimates that there are over 300,000 OTC drug products currently available.9 Although both prescription and OTC products meeting the FDA’s definition of a drug are regulated by the FDA, they are distinguishable based upon their safety, the need for supervision, and the risk of self-treatment. If, in the FDA’s judgment, a drug cannot be used safely without adequate supervision by a healthcare practitioner, it will be approved for prescription only. Specifically, the FDCA states that a drug must be dispensed by prescription if, ‘‘because of its toxicity or other potentiality for harmful effect, or the method of its use, or the collateral measures necessary to its use, [it] is not safe for use except under the supervision of a practitioner licensed by law to administer such drug.”9 The FDA also requires that OTC drugs have a standardized label format and content that includes the name of the active ingredient(s), indication, and directions.10
Instead of reviewing the vast number of individual OTC drug products, the FDA reviews the active ingredients and the labeling of over 80 therapeutic classes of drugs, for example, analgesics or antacids.9 For each category of OTC drug, a monograph is developed and published in the Federal Register. OTC drug monographs are likened to a “recipe book” covering acceptable ingredients, doses, formulations, and labeling and define the safety, effectiveness, and labeling of OTC active ingredients. Once a final monograph is implemented, companies can make and market an OTC product that conforms to a final monograph without the need for FDA preapproval and review. The NDA process reviews those products that do not conform to an existing monograph. A drug company may also petition the FDA to change a final monograph to include additional ingredients or to modify labeling.9 While compliance with the monograph removes the necessity of providing safety data for individual products, OTC products are nev ertheless subject to postmarketing safety review.
When the FDA is considering the approval of a drug for the OTC market, it expects that the drug will generally have these characteristics11:
- It can be adequately labeled such that the consumer can self-diagnose, self-treat, and self manage the condition being treated.
- No healthcare practitioner is needed for the safe and effective use of the product.
- The safety margin adequately ensures that the benefits of OTC availability outweigh its risks.
- The drug has a low potential for misuse and abuse.
In making its determination, the FDA relies on the recommendations of the Nonprescription Drugs Advisory Committee, which reviews and evaluates available data concerning the safety and effectiveness of OTC drugs; however, the committee’s recommendation is not binding on the FDA.12 A manufacturer can also request that a drug that has been approved for distribution only on prescription be switched to OTC.11 In general, the manufacturer submits information about the drug’s safety, including experience gained while it was a prescription drug; that the drug is appropriate for self-administration; and that the labeling can be read, understood, and followed by a consumer without guidance from a healthcare professional. More than 100 OTC ingredients, strengths, or indications are on the market today that were available only by prescription fewer than 40 years ago.
Dietary Supplements
Dietary supplements are also sold without a prescription, but the FDA regulates dietary supplements under a set of regulations that differ from those covering conventional foods and OTC or prescription drug products. Supplements are subject to the Dietary Supplement Health and Education Act of 1994 (DSHEA), rather than the FDCA.13 Under DSHEA, a dietary supplement is defined as a product intended to supplement the diet that contains one of the following ingredients: “vitamin; mineral; herb or other botanical; amino acid; dietary substance for use by man to supplement the diet by increasing the total dietary intake; or a concentrate, metabolite, constituent, extract, or combination of the preceding substances,” which are marketed in nonconventional food forms such as tablets, capsules, softgels, gelcaps, powders, and liquids.13 Therefore, they are neither foods nor drugs.
The FDA acknowledges that Congress intended for DSHEA to provide broad access to dietary supplements and considered supplements marketed prior to the passage of DSHEA to be generally safe.14 The FDA permits these established products to be marketed freely, similar to conventional foods such as fresh fruits and vegetables.15 If the supplement contains a New Dietary Ingredient (NDI), DSHEA requires the manufacturer to notify the FDA at least 75 days prior to marketing and to include in the notification the manufacturer’s basis for concluding that the dietary supplement containing the ingredient “will reasonably be expected to be safe.”15 However, unlike the case with drugs, the manufacturer is not required to wait for a safety determination from the FDA before marketing the product. Although premarketing approval is not required, the FDA has the authority to initiate proceedings to remove a substance from the market if it poses a “significant or unreasonable” risk to consumers or if it is considered to otherwise be adulterated or contain inaccurate labeling.14
The FDA issued new draft guidance in 2016 regarding NDIs that was designed to improve the rate of compliance and quality of notifications.16 The FDA noted that more than 5,000 new dietary supplement products enter the marketplace each year, compared with 4,000 products on the market in 1994, when DSHEA was enacted. The draft guidance includes information on what qualifies as an NDI, when notification is required, and the types of data recommended by the FDA when evaluating safety.15,16 One commentator has noted that this may pose a conflict between an assertion of the FDA’s granted regulatory authority over supplements and the FDA overstepping its bounds and treating supplements as drugs, which would contravene Congress’ intent in passing DSHEA.15
An important distinction between drugs and dietary supplements is the claims that can be made. Supplements may not make a disease claim (i.e., supplements may not be marketed or promoted as intending to treat, diagnose, prevent, or cure diseases), or they would be treated as drugs.17 This means that supplements may not claim to “reduce pain” or “treat heart disease” and may not incorporate a drug name by reference (e.g., Herbal Prozac). They may, however, make structure-function claims. A structure-function claim as it pertains to supplements describes “the role of a nutrient or dietary ingredient intended to affect the normal structure or function of the human body; for example, ‘calcium builds strong bones.’” In addition, a claim may characterize the means by which a nutrient or dietary ingredient acts to maintain such structure or function (for example, “fiber maintains bowel regularity” or “antioxidants maintain cell integrity”) and may make claims of general well being.17
Structure-function claims are not preapproved by the FDA but must be truthful and not misleading, and the manufacturer must notify the FDA of the claim no later than 30 days after marketing the product. The now-familiar disclaimer that the FDA has not evaluated the claim must also accompany the claim. The disclaimer must also state that the dietary supplement product is not intended to “diagnose, treat, cure or prevent any disease,” or it would be considered a drug.14,15 The distinction between a permissible structure-function claim and an impermissible disease claim is often blurry.
Although supplements do not require premarket approval by the FDA, the FDA can take action to remove products from the market, but the agency must first establish that such products are adulterated (e.g., that the product is unsafe) or misbranded (e.g., that the labeling is false or misleading).14 The FDA relies on postmarket surveillance to identify a risk to the public and has the burden of proving that a particular product is not safe. As a result, the FDA has been slow to remove dietary supplements from the market, taking years in some cases to assemble adequate scientific data to prove harm and complete the regulatory hurdles.18
An example of the protracted process is the FDA’s action to ban the stimulant dimethylamylamine (DMAA), an amphetamine-like product found in geraniums.19 DMAA was contained in supplements promising weight loss, muscle building, and performance enhancement. Like most stimulants, it can elevate blood pressure and lead to cardiovascular problems. The FDA received 86 reports of serious illness or death associated with DMAA and issued letters to manufacturers in 2013 that products containing this ingredient are illegal, which, in the FDA’s view, was “the quickest way at FDA’s disposal to halt the further distribution of dietary supplements.”19 One company failed to comply, and as of 2017, the FDA’s order was still being litigated.
Consumers are largely unaware that dietary supplements are not required to obtain FDA approval and that there is no requirement for manufacturers to prove safety. While there are many excellent and safe dietary supplements available, it is incumbent on pharmacists to remind consumers that while the rate of adverse events is small compared with rates for prescription drugs, “natural” does not automatically equate to safety, and that the FDA monitors natural products for toxicity as well as hidden ingredients, including on occasion prescription drugs, which may pose a risk of toxicity.15 The FDA modified its regulations in 2007 to require that supplements follow good manufacturing practices (GMPs) to establish a comprehensive system of process controls in order to consistently meet established specifications for identity, purity, strength, and composition; it also requires that they have been manufactured, packaged, and held under conditions to prevent adulteration. Some supplements also exhibit various seals on their labels (e.g., USP, NF, UL, ConsumerLab). Generally, these seals indicate that the product meets certain standards for purity and adulteration, but do not provide an endorsement of therapeutic value. The seals represent a voluntary compliance by the manufacturer, which pays for the testing that takes place at different intervals (one to six times per year) on samples provided by the manufacturer or purchased from stores, depending on the organization providing the seal. Only a small percentage of products undergo testing and receive seals.
Homeopathic Drugs
Homeopathic drugs occupy a special position in the regulatory framework. Unlike dietary supplements, the FDA can substantially regulate homeopathic products because the FDCA allows them to make a therapeutic claim.20 These drugs, however, are not subject to the same level of FDA scrutiny as allopathic drugs.
Homeopathy was established in 1796, and most popular homeopathic medicines have been in the marketplace in the U.S. for 50 years or more.21 However, the homeopathy industry is small compared with the OTC drug, prescription drug, and dietary supplement industries in terms of revenues, advertising, and the number of marketed products.21 Although there are more than 7,000 homeopathic medicines registered with the FDA, it is estimated that only about 1,000 are marketed on a routine basis. Estimates of product sales in the U.S. estimate a growth rate of roughly 5% per year. The majority of homeopathic medicines are used to treat cough, cold and flu, muscle pain, and children’s ailments and represent less than 3.5% of all OTC drug products typically found in drugstore chains.21
When the FDCA was enacted in 1938, it recognized drugs and standards published in the Homeopathic Pharmacopeia of the United States (HPUS) as official. Homeopathic drugs are defined as “any drug labeled as being homeopathic which is listed in the Homeopathic Pharmacopeia of the United States (HPUS), an addendum to it, or its supplements.”20,22
The HPUS consists of monographs of official homeopathic drugs.23 The monographs list complete identifying data for the drug, as well as specific manufacturing standards. The HPUS is prepared by a nongovernmental organization, the Homeopathic Pharmacopoeia Convention of the United States (HPCUS), composed of scientists and clinicians who are trained in the medical specialty of homeopathic medicine.23,24 HPCUS is a standard-setting organization focusing on the regulatory approval of official homeopathic drug products and the development and publication of general practices and standards. The HPUS has been in continuous publication since 1841, and 1,286 official homeopathic drug products are currently recognized.23,24
Monographs, which are evaluated by a review committee composed primarily of scientists and pharmacists, focus on technical criteria. These include substance characterization, quality analysis and controls, assay techniques, and reference reagents, as well as the technical aspects of drug production. An accepted monograph is published in journals related to the professional practice of homeopathic medicine for a 90-day comment period.23,24
Any drug listed in an HPUS monograph is considered official and requires no further documentation by the manufacturer to be sold.22 A nonlisted drug is considered unofficial and requires the manufacturer to provide sufficient clinical data to the FDA in order to make a determination as to whether the drug is, in fact, homeopathic.22 If a drug is recognized in both the United States Pharmacopeia and the HPUS, it is subject to the requirements of the USP unless it is labeled and offered for sale as a homeopathic drug, in which case it is subject to the provisions of the HPUS.22 Homeopathic drug products must contain diluents commonly used in homeopathic pharmaceuticals. If a product contains homeopathic ingredients in combination with nonhomeopathic active ingredients, it is not considered to be a homeopathic drug product. Like nonhomeopathic products, if it is offered for the cure, mitigation, prevention, or treatment of disease conditions, it is regarded as a drug within the meaning of the FDCA.
Homeopathic drugs, like allopathic compounds, are divided into prescription and OTC categories and follow similar criteria for these designations. Essentially, if a homeopathic product is intended solely for a selflimiting disease that can be diagnosed and treated by a nonhealthcare professional, it may be marketed as an OTC product. If not, it must be offered only as a prescription product.22
The original 1938 FDCA focused on the safety, rather than efficacy, of new drugs, and products listed in the HPUS were considered to have met the new standards.20 The FDCA was modified in 1962 by the Kefauver-Harris Amendments, which mandated proof of efficacy of conventional drugs through “well controlled investigations,” but the status of homeopathic drugs was unchanged.20 Homeopathic products were also excluded from the FDA’s Drug Efficacy Study Implementation process, in which all drugs approved between 1938 and 1962 were retrospectively evaluated, and the FDA’s review of OTC drugs. In the latter case, homeopathic drugs were to be reviewed “at a later time” but have not been to date.20 In addition, homeopathic drug products historically have carried little or no labeling for the consumer.
It is apparent that even after the enactment of the 1962 amendments to the FDCA, homeopathic drugs remained in an ambiguous regulatory position. FDA action has traditionally been largely based on institutional understanding and informal agreements between the FDA and members of the homeopathic industry.24 As a result, there was a lack of consistency and clarity in applying regulatory control that left the industry without clear guidelines.24 In addition, all homeopathic drugs were prescription only.22,24
In recognition of the expansion of the homeopathic drug market, the FDA issued a Compliance Policy Guide (CPG) in 1988 that provided guidance on regulation and delineated the conditions under which homeopathic drugs could be marketed.22 The CPG established criteria for allowing homeopathic drugs to be sold OTC. These criteria closely aligned with those of other OTC products, permitting OTC sales if the drugs had low toxicity and were offered for self-limiting and self-diagnosable conditions. The CPG also altered the labeling, requiring drugs to have at least one indication on the label and a package insert (if prescription), a list of ingredients, and directions for use.20,24 In addition, conformity with GMPs was required, and the CPG alerted regulators and the industry that promotion of drugs beyond “recognized or customary practice” would be scrutinized for possible health fraud.20,22
In April 2015, the FDA published a notice seeking comments from stakeholders about drugs labeled as homeopathic and its regulatory framework.25 The FDA’s intent was to review its regulation of homeopathic products. In the announcement, the FDA recognized that there have been many changes since its 1988 Compliance Guide. The FDA noted that over that 27-year period, there was a large growth in the homeopathic market, especially in the OTC area, growing from a multimillion-dollar market to sales of almost $3 billion in 2007. The FDA also commented “(m)any drugs labeled as homeopathic are sold OTC in major retail stores and are often marketed as natural, safe, and effective alternatives to other prescription and nonprescription products.”25 The FDA expressed its intent to evaluate its current enforcement policies of homeopathic drugs from “scientific, risk, and process perspectives” and to solicit opinions about whether its limited oversight is appropriate to protect and promote public health and whether and how its current enforcement policies needed to be adjusted to conform with changes in the homeopathic product marketplace.
Part of the FDA’s safety concern derived from data it presented from the 2012 American Association of Poison Control Center Annual Report, which indicated that there were 10,311 reported poison-exposure cases related to Homeopathic Agents, with 8,788 of these cases occurring in children aged 5 years and younger.21 Of the reported cases, 697 required treatment in a healthcare facility.
At the hearing, the Consumer Healthcare Products Association (CHPA), a trade association representing manufacturers of OTC drugs and supplements, challenged this interpretation, noting that the homeopathic products have a very favorable safety profile and that the analysis of the poison center data shows a low rate of overall exposure, with most exposures categorized as little or no effect.26 The CHPA did support regulatory standards for OTC homeopathic drugs that would be similar to those for OTC allopathic drugs, including facility registration and inspection, standardized labeling, and serious adverse event reporting. Clearly, homeopathic drugs have shown up on the FDA’s radar screen.
A few months later, the Federal Trade Commission (FTC) also turned its attention to homeopathic products.27 (The FTC has jurisdiction over advertising of OTC [allopathic and homeopathic] products. As a result of a 1971 Memorandum of Understanding between the two federal agencies, the FDA focuses on OTC product labeling and the FTC focuses on OTC product advertising and marketing.27 The FDA is responsible for both labeling and advertising of prescription drugs.) The FTC was concerned that the FDA’s requirement that labeling for homeopathic drugs include an indication for use, even when the product has not demonstrated any efficacy for that indication, creates a potential conflict with the FTC’s requirement that health-related claims be substantiated by “competent and reliable scientific evidence.”
Following a public hearing in September 2015, the FTC concluded that there is “no basis under the FTC Act to treat OTC homeopathic drugs differently than other health products.” Furthermore, the FTC concluded that efficacy and safety claims for OTC homeopathic drugs should be held to the same truth-in advertising standards as other products claiming health benefits.27 (The Federal Trade Commission Act empowers the agency to, among other things, “prevent unfair methods of competition and unfair or deceptive acts or practices in or affecting commerce” and “prescribe rules defining with specificity acts or practices that are unfair or deceptive, and establishing requirements designed to prevent such acts or practices.”28)
As a result of the hearing, the FTC decided “to address concerns that many consumers are likely being misled by current (homeopathy) marketing claims.” In the FTC’s opinion, “Efficacy claims for traditional OTC homeopathic products are only supported by homeopathic theories and homeopathic provings, which are not accepted by most modern medical experts and do not constitute competent and reliable scientific evidence that these products have the claimed treatment effects. For these reasons, the vast majority of OTC homeopathic drugs lack adequate substantiation for their efficacy claims.”28
In accordance with their statutory obligations, the FTC issued an enforcement-policy statement that marketers of OTC homeopathic products must either have adequate support for their claims of efficacy or effectively communicate that there is a lack of scientific evidence to support the claims and that the claims are based on theories that date back to the 1700s and are not accepted by most modern medical experts. However, the FTC added that a nonmisleading efficacy claim could be possible and further stated that a supplier could include an indication not supported by scientific evidence as long as they effectively communicate the basis for the claim.28
Drug Regulation and the Role of the Pharmacist
The FDA is responsible for the oversight of prescription drugs, OTC drugs, homeopathic drugs, and dietary supplements. However, the nature of oversight and the regulatory requirements differ for each group of products (see TABLE 1). Prescription drugs require the most stringent evidence of safety and efficacy and receive the most rigorous oversight from the FDA, including preapproval before marketing. Prescription, OTC, and homeopathic products all fall within the FDA’s definition of a drug and require an indication in their labeling. Dietary supplements fall outside of the definition of a drug and not only do not require an indication on the label, but are prohibited from doing so.
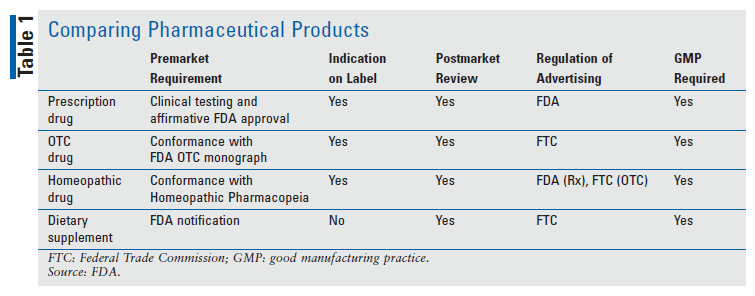
As described above, regulatory changes are being considered and enacted to clarify the status and regulatory oversight of medicinal agents. In particular, the relatively lax oversight of homeopathic products and supplements has caught the attention of the FDA and the FTC. Further regulatory changes are also being considered by Congress, including a bill that would grant the FDA mandatory recall authority over drugs instead of voluntary compliance and protracted legal impediments.29
One troubling area where pharmacists can make a significant contribution is consumers’ and healthcare practitioners’ lack of knowledge about the regulation of alternative medicinal products. Surveys have shown, for example, that 60% of healthcare professionals did not know what DSHEA is, what it means, how it affects them as practitioners, or how it affects the consumer of supplements.15 Another survey found that at least one-third of physicians were not aware that dietary supplements do not require FDA approval or submission of safety and efficacy data before being marketed.30 A comparable percentage of physicians believed that there are regulations that ensure supplement quality. Perhaps more troubling, most physicians were not aware that serious adverse events due to the use of supplements should be reported through the FDA MedWatch program.
Consumers also have information gaps in their understanding of supplement oversight. Surveys have shown that more than half of consumers are unaware that dietary supplements do not need to be approved by the FDA, while 63% are not aware that advertisements for supplements do not need to be preapproved.31 Significantly, a study found that educating consumers about DSHEA made them more skeptical about the safety and effectiveness of dietary supplements, suggesting that increased consumer awareness is beneficial.32 Pharmacists are in a unique position to address these deficiencies and improve patients’ awareness and effective use and selection of the many products found on pharmacy shelves.
REFERENCES
- U.S. Food and Drug Administration. Regulated Product. www.fda.gov/ForIndustry/ImportProgram/ImportBasics/RegulatedProducts/default.htm.
- U.S. Food and Drug Administration. FDA’s New Regulations for E-Cigarettes, Cigars, and All Other Tobacco Products. www.fda.gov/tobaccoproducts/labeling/rulesregulationsguidance/ucm394909.htm.
- Merrill RA. Regulation of drugs and devices: an evolution. Health Affairs. 1994;13(3):47-69.
- Van Norman GA. Drugs, devices, and the FDA: part 1: an over view of approval processes for drugs. JACC Basic Trans Sci. 2016;1:170-179.
- Juhl RP. Prescription to over-the-counter switch: a regulatory perspective. Clin Ther. 1998;20(supp. 3):C111-C117.
- U.S. Food and Drug Administration. Drug Applications for Over the-Counter (OTC) Drugs. www.fda.gov/drugs/developmentapprovalprocess/howdrugsaredevelopedandapproved/approvalapplications/over-the-counterdrugs/default. htm.
- U.S. Food and Drug Administration. The FDA’s Drug Review Process: Ensuring Drugs Are Safe and Effective. www.fda.gov/drugs/resourcesforyou/consumers/ucm143534.htm.
- U.S. Food and Drug Administration. Unapproved Prescription Drugs: Drugs Marketed in the United States That Do Not Have Required FDA Approval. www.fda.gov/Drugs/GuidanceComplianceRegulatoryInformation/EnforcementActivitiesbyFDA/ SelectedEnforcementActionsonUnapprovedDrugs/default.htm.
- U.S. Food and Drug Administration. Drug Applications for Over the-Counter (OTC) Drugs. www.fda.gov/drugs/developmentapprovalprocess/howdrugsaredevelopedandapproved/approvalapplications/over-the-counterdrugs/default.htm.
- U.S. Food and Drug Administration. Over-The-Counter Human Drugs; Labeling Requirements. Fed Regist. 2009;64(51):13254-13303.
- Michele TM. Regulatory Approaches for Prescription to OTC Switch. www.fda.gov/downloads/Drugs/NewsEvents/UCM454815.pdf.
- U.S. Food and Drug Administration. Nonprescription Drugs Advisory Committee. www.fda.gov/advisorycommittees/committeesmeetingmaterials/drugs/nonprescriptiondrugsadvisorycommittee/default.htm.
- U.S. Food and Drug Administration. Dietary Supplements. www.fda.gov/food/dietarysupplements.
- Henney JE. Implementation of the Dietary Supplement Health and Education Act (DSHEA) of 1994. www.fda.gov/newsevents/testimony/ucm115082.htm.
- Stohs SJ, Preuss HG. What healthcare professionals should know about the regulation and safety of dietary supplements. J Am Coll Nutr. 2017;36(4):306-309.
- U.S. Food and Drug Administration. Dietary Supplements: New Dietary Ingredient Notifications and Related Issues: Guidance for Industry. www.fda.gov/downloads/Food/GuidanceRegulation/GuidanceDocumentsRegulatoryInformation/UCM515733.pdf.
- U.S. Food and Drug Administration. Structure-Function Claims. www.fda.gov/Food/IngredientsPackagingLabeling/LabelingNutrition/ucm2006881.htm.
- Wallace TC. Twenty years of the Dietary Supplement Health and Education Act—how should dietary supplements be regulated? J Nutr. 2015;145(8):1683-1686.
- U.S. Food and Drug Administration. Stimulant Potentially Dangerous to Health, FDA Warns. www.fda.gov/ForConsumers/ConsumerUpdates/ucm347270.htm.
- Podolsky SH, Kesselheim AS. Regulating homeopathic products—a century of dilute interest. N Engl J Med. 2016;374:201-203.
- U.S. Food and Drug Administration. Homeopathic Product Regulation: Evaluating FDA’s Regulatory Framework After a Quarter Century. www.fda.gov/Drugs/NewsEvents/ucm430539.htm.
- U.S. Food and Drug Administration. CPG Sec. 400.400 Condi tions Under Which Homeopathic Drugs May be Marketed. www.fda.gov/iceci/compliancemanuals/compliancepolicyguidancemanual/ucm074360.htm.
- Borneman JP, Field RI. Regulation of homeopathic drug products. Am J of Hlth-System Pharm. 2006;63(1):86-91.
- The Homœopathic Pharmacopœia of the United States. www. hpus.com/legal-status.php#.
- U.S. Food and Drug Administration. Homeopathic Product Regulation: Evaluating the Food and Drug Administration’s Regulatory Framework After a Quarter-Century; Public Hearing. Fed Regist. 2015;80(59):16327-16329.
- Consumer Healthcare Products Association. Homeopathic Product Regulation. www.fda.gov/downloads/Drugs/NewsEvents/UCM443486.pdf.
- U.S. Federal Trade Commission. Homeopathic Medicine & Advertising Workshop Report. www.ftc.gov/system/files/documents/reports/federal-trade-commission staff-report-homeopathic-medicine-advertising-workshop/p114505_ otc_homeopathic_medicine_and_advertising_workshop_report.pdf.
- U.S. Federal Trade Commission. Federal Trade Commission Act. www.ftc.gov/enforcement/statutes/federal-trade-commission-act.
- DeLauro Introduces Bill to Give FDA Mandatory Recall Authority over Drugs. https://delauro.house.gov/media-center/press-releases/delauro-introduces-bill-give-fda-mandatory-recall-authority-over drugs.
- Ashar BM, Rice TN, Sisson SD. Physicians’ understanding of the regulation of dietary supplements. Arch Intern Med. 2007;167:966-969.
- Ashar BM, Miller RG, Pichard CP, et al. Patients’ understanding of the regulation of dietary supplements. J Community Health. 2008;33(1):22-30.
- Dodge T, Litt D, Kaufman A. Influence of the Dietary Supplement Health and Education Act on consumer beliefs about the safety and effectiveness of dietary supplements. J Hlth Comm. 2011;16(3):230-244.