The FDA Accelerated Drug Approval Program Explained
RELEASE DATE
October 1, 2023
EXPIRATION DATE
October 31, 2025
FACULTY
Donna M. Lisi, PharmD, BCPS, BCACP, BCGP, BCPP,
BCMTMS
Independent Clinical Consultant Pharmacist
Somerset, New Jersey
FACULTY DISCLOSURE STATEMENTS
Dr. Lisi has no actual or potential conflicts of interest in relation to this activity.
Postgraduate Healthcare Education, LLC does not view the existence of relationships as an implication of bias or that the value of the material is decreased. The content of the activity was planned to be balanced, objective, and scientifically rigorous. Occasionally, authors may express opinions that represent their own viewpoint. Conclusions drawn by participants should be derived from objective analysis of scientific data.
ACCREDITATION STATEMENT
 Pharmacy
Pharmacy
Postgraduate Healthcare Education, LLC is accredited by the Accreditation Council for Pharmacy Education as a provider of continuing pharmacy education.
UAN: 0430-0000-23-103-H03-P
Credits: 2.0 hours (0.20 ceu)
Type of Activity: Knowledge
TARGET AUDIENCE
This accredited activity is targeted to pharmacists. Estimated time to complete this activity is 120 minutes.
Exam processing and other inquiries to:
CE Customer Service: (800) 825-4696 or cecustomerservice@powerpak.com
DISCLAIMER
Participants have an implied responsibility to use the newly acquired information to enhance patient outcomes and their own professional development. The information presented in this activity is not meant to serve as a guideline for patient management. Any procedures, medications, or other courses of diagnosis or treatment discussed or suggested in this activity should not be used by clinicians without evaluation of their patients' conditions and possible contraindications or dangers in use, review of any applicable manufacturer's product information, and comparison with recommendations of other authorities.
GOAL
To educate pharmacists about the FDA's Accelerated Approval (AA) program and to describe its strengths and limitations compared with the traditional drug and biological approval process.
OBJECTIVES
After completing this activity, the participant should be able to:
- Describe the history and purpose of the FDA's AA program.
- Define the role and types of surrogate endpoints used in the FDA's AA program.
- Identify the major drug classes and the classification of the stage of status approval for drugs/biologicals and indications in the AA program.
- List limitations of the FDA's AA process and possible strategies to improve the AA program.
ABSTRACT: Created in 1992 in response to the AIDS/HIV epidemic, the FDA's Accelerated Approval (AA) program was designed to allow for the approval of drugs and biologicals for serious conditions using surrogate endpoints instead of clinical outcomes as are used in traditional drug approvals. Surrogate endpoints are of two types: validated and reasonably likely. Since these endpoints do not represent clinical outcomes, it is essential that confirmatory trials be performed to determine the therapeutic benefit of drugs or biologicals approved via the AA pathway. There are numerous limitations to the use of surrogate endpoints. Pharmacists need to be aware of the opportunities of the AA program as well as its weaknesses, which include drugs and biologicals coming to market with less vigorous data supporting their use compared with the traditional drug approval process.
To expedite the approval of drugs and biologicals to treat serious conditions, the FDA has developed four distinct approaches to promote the rapid approval of medications. These approaches include priority review, a process that indicates the FDA will act upon the new drug or biological application within 6 months; fast track, which is a process designed to facilitate the development and expedite the review of drugs to treat serious conditions and fill an unmet medical need; breakthrough therapy, a process designed to expedite the development and review of drugs that may demonstrate substantial improvement over available therapy; and Accelerated Approval (AA), a regulatory approval process that allows for drugs or biologicals indicated for serious conditions that fulfill an unmet medical need to be approved based on a surrogate endpoint.1 This article will discuss the AA program.
HISTORY OF THE AA PROGRAM
Regulations governing the AA pathway were passed in 1992 in response to the AIDS epidemic. The goal of the program was to allow drugs for serious conditions for which there was an unmet need, such as AIDS or HIV, to be approved based on a surrogate endpoint, which could result in speedier drug approvals. In 2012, Congress codified the AA program by the passage of the FDA Safety and Innovation Act (FDASIA). Section 901 of the FDASIA amended the Food, Drug, and Cosmetic Act (FDCA) so that drugs in the AA program could be approved based on either a surrogate endpoint or an intermediate clinical endpoint that is reasonably likely to predict clinical benefit. Support for the use of a proposed surrogate or intermediate clinical endpoint should be based on scientific support from epidemiological, therapeutic, or pathophysiologic data and clinical trials.2-4
During the period of 1992 to 2001, 40% of 40 product-indication pairs with AA were indicated for HIV/AIDS. In these early days of the program, only 30% were indicated for cancer, and another 30% were approved for other conditions. However, a drastic shift took place over time as HIV has become more of a chronic condition, with 93.3% of product-indication pairs granted AA for the treatment of cancer in 2020.5
LEGISLATION REGULATING THE AA PROGRAM
According to the FDCA, a biomarker refers to "a characteristic (such as a physiologic, pathologic, or anatomic characteristic or measurement) that is objectively measured and evaluated as an indicator of normal biologic processes, pathologic processes, or biological responses to a therapeutic intervention; and includes a surrogate endpoint."6 A "surrogate endpoint" is defined as "a marker, such as a laboratory measurement, radiographic image, physical sign, or other measure, that is not itself a direct measurement of clinical benefit, and either (a) is known to predict clinical benefit and could be used to support traditional approval of a drug or biological product; or (b) is reasonably likely to predict clinical benefit and could be used to support the AA of a drug or biological product."6
Under the AA process, a drug may be approved if it is for a serious or life-threatening disease or condition, "upon a determination that the product has an effect on a surrogate endpoint that is reasonably likely to predict clinical benefit, or on a clinical endpoint that can be measured earlier than irreversible morbidity or mortality, that is reasonably likely to predict an effect on irreversible morbidity or mortality or other clinical benefit, taking into account the severity, rarity, or prevalence of the condition and the availability or lack of alternative treatments." The evidence that is needed under the AA process supports "that an endpoint is reasonably likely to predict clinical benefit" and may include epidemiological, pathophysiological, therapeutic, pharmacologic, or other evidence developed using biomarkers or other scientific methods or tools.6
The 21st Century Cures Act mandates that the FDA publish a list of surrogate endpoints used as a basis to approve or license a drug or biological product under both accelerated and traditional approval provisions.7,8
CLASSIFICATION OF MEDICATION STATUS
Classification of the status of drugs approved in the AA process fall into the following categories9:
Ongoing: Drugs/biologicals that have indications with postmarketing requirements for ongoing clinical trials to verify their clinical benefit. As of August 14, 2023, six pharmaceuticals with infectious disease indications excluding vaccines; six vaccines; 27 drugs/biologicals for nonmalignant hematologic, neurologic, and other disorders; and 66 cancer agents with AAs fell within this category. The FDA has established a Postmarketing Requirements and Commitments website that describes the confirmatory studies required to fulfill the obligations of the AA status (www.accessdata.fda.gov/scripts/cder/pmc/index.cfm?resetfields=1).
Verified Clinical Benefit: Drugs/biologicals that have received AA for indications for which postmarketing trials have verified the pharmaceutical's clinical benefit and for which traditional approval has been subsequently granted for the specific indication. As of August 14, 2023, 32 pharmaceuticals with infectious disease indications excluding vaccines; 12 vaccines; 20 drugs/ biologicals for nonmalignant hematologic, neurologic, and other disorders; and 94 cancer agents that initially received AA status fell into this category have since acquired full, indication-specific, traditional FDA approval status.
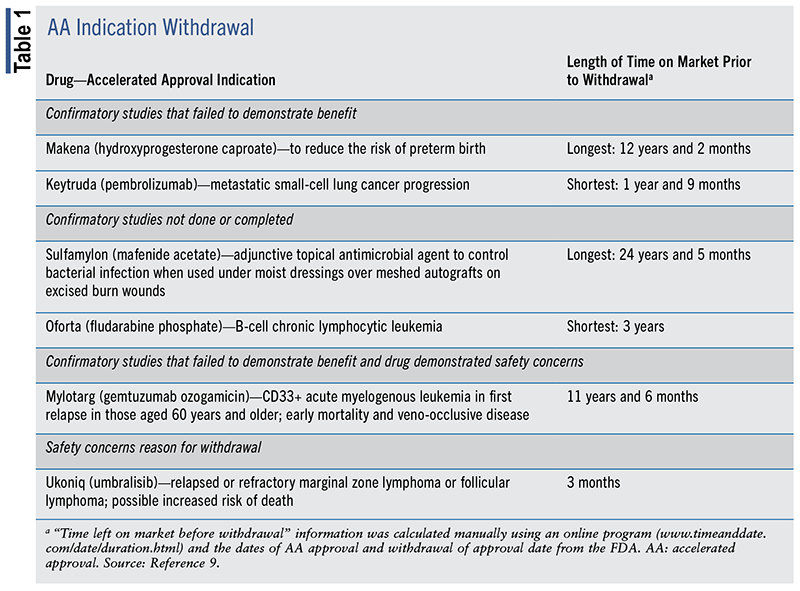
Withdrawn: Drugs/biologicals granted AA with postmarketing requirements that have subsequently been withdrawn and, therefore, are no longer FDA-approved.9 (See TABLE 1.) Withdrawal of AA status is due to either negative confirmatory test results or failure to complete confirmatory testing, as required. As of August 14, 2023, nine pharmaceuticals with infectious disease indications; four drugs/biologicals for nonmalignant hematological, neurological, and other disorders; and 26 cancer agents had their AA status withdrawn and are no longer FDA-approved medications. Twenty AA indications have been voluntarily withdrawn by sponsors since December 2020.10
Other: AAs for malignant hematology and oncology indications that have been granted approval status as support-care products. As of August 14, 2023, seven drugs/biologicals fell within this category.
INDICATIONS OF MEDICATIONS IN THE AA PROGRAM
Indications for which drugs or biologicals are currently under investigations (considered "ongoing") in the AA program as of August 14, 2023, include cancer, infectious diseases (nonvaccine and vaccine indications); nonmalignant hematologic, neurologic, and other indications (i.e., intermediate- or high-risk primary or secondary myelofibrosis with thrombocytopenia, sickle cell disease, reversal of anticoagulation needed due to life-threatening or uncontrolled bleeding, methemoglobinemia); neurologic indications (e.g., Alzheimer's disease, Duchenne muscular dystrophy, symptomatic neurogenic orthostatic hypotension); and other (i.e., proteinuria in primary immunoglobulin A nephropathy, active cerebral adrenoleukodystrophy, achondroplasia, Fabry disease with amenable galactosidase alpha gene variant, primary biliary cholangitis with ursodeoxycholic acid [udca] who had inadequate response to udca or used as monotherapy if unable to tolerate udca, Hunter syndrome, and immune complications from smallpox vaccination).9
CLINICAL TRIAL ENDPOINTS IN THE TRADITIONAL VERSUS AA PROCESS
In the traditional drug approval process, a sponsor must demonstrate that a drug is safe and effective. Effectiveness is based on substantial evidence, which is defined as evidence consisting of adequate and well-controlled investigations, including clinical investigations, by experts qualified by scientific training and experience to evaluate the effectiveness of the drug as prescribed, recommended, or suggested in the labeling.11 Clinical outcomes can include, for example, an improvement in symptoms or a decrease in mortality or can serve as tools to assess whether the benefits of the new therapy outweigh risks. Traditional drug approval trials are usually randomized, placebo-controlled studies.3
The traditional drug approval process can take 12 to 15 years.12 The AA program has led to drugs and biologicals being available a median of 3.1 years earlier.13,14
The AA process involves the use of surrogate endpoints and is employed in situations where it may take a long time to achieve a clinical outcome (e.g., neurologic conditions) or when the clinical benefit associated with improvement in the surrogate endpoint is well understood (e.g., hypertension).3,9
There may also be situations where conducting a clinical endpoint study using a randomized, placebo-controlled methodology may be unethical because the placebo group would be untreated.3
FDA-APPROVED SURROGATE ENDPOINTS
Surrogate endpoints that have undergone extensive testing and are supported by a clear mechanistic rationale and clinical data providing strong evidence that an effect on the surrogate endpoint predicts a specific clinical benefit are called validated surrogate endpoints. On the other hand, "reasonably likely surrogate endpoints" are endpoints that are supported by strong mechanistic and/or epidemiologic rationale but the amount of clinical data available is not sufficient to show that they are a validated surrogate endpoint. It is extremely important that postmarketing confirmatory trials are completed to determine if the reasonably likely surrogate endpoint predicts clinical benefit.3
Among the types of biomarkers are molecular, histologic, radiographic, or physiologic characteristics. Biomarker classes include those that indicate efficacy response or safety or are diagnostic. Uses of biomarkers vary from enrichment of study protocols to being predictive or prognostic of a clinical outcome.3
Biomarkers are developed through the Biomarker Qualification Program and are distinct from clinical outcome assessments, which are measured using a report generated by a clinician, patient, nonclinician observer, or a performance-based assessment.3
TABLE 2 and TABLE 3 contain a list of the approved surrogate endpoints used in adult and pediatric research, respectively. These surrogate endpoints serve as a proxy for clinical efficacy.
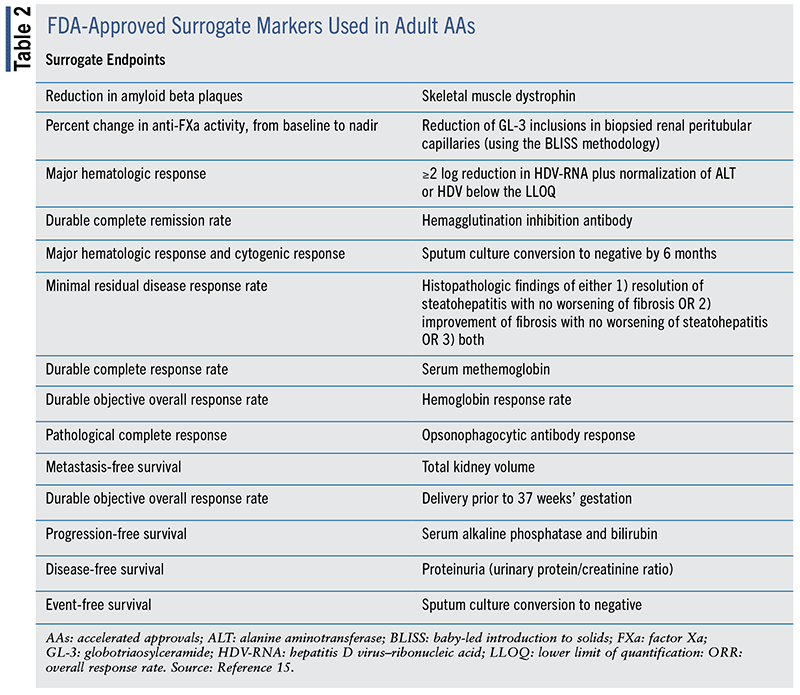

Surrogate endpoints are used in both traditional approval and AA. For surrogate endpoints employed solely in traditional drug approvals, refer to the FDA Surrogate Endpoint Table at www.fda.gov/drugs/development-resources/table-surrogate-endpoints-were-basis-drug-approval-or-licensure. Traditional drug approval trials also include clinical outcomes.
While the FDA has indicated that a particular surrogate endpoint may be acceptable in a particular drug or biological development program, its use is context-dependent and needs to be considered on a case-by-case basis. Among the factors that need to be taken into account are the disease state for which the treatment is intended, the study population, the therapeutic mechanism of the drug as it relates to the surrogate endpoint, and the availability of current treatments.15
For drugs or biologicals in development for cancer, endpoints that are based on changes in tumor burden need to take into consideration the disease being treated, effect size, effect duration, residual uncertainty, and benefits of other available therapies.15
Surrogate endpoints that are approved for use in clinical trials but have not been utilized as of February 28, 2022 (the last time that the FDA updated the list) include event-free survival in the treatment of patients with breast cancer or neuroblastoma; use of ≥2 log reduction in hepatitis D virus (HDV)-RNA plus normalization of alanine aminotransferase or HDV below the lower limit of quantification for hepatitis D; total kidney volume in polycystic kidney disease in adults; and use of urinary protein/creatinine ratio in primary glomerular diseases associated with significant proteinuria in pediatric patients.15
BENEFITS OF THE AA PROGRAM
Rare, Serious, Life-Threatening Diseases
The AA program offers hope for the development of treatments for rare diseases. Of the nearly 7,000 known rare diseases, many are life-threatening or life-limiting and have a genetic basis with manifestations occurring in early childhood. Due to the rarity of these conditions, it is often not feasible to conduct large randomized, controlled trials.16
A review of FDA data found that from 2008 to 2021 among the drug-indication pairs approved through the AA program, 70 of 82 (85.4%) had an orphan designation; of those, 55 (78.6%) were oncology drugs.17
Quick Conversion to Traditional FDA Approval
An example of an AA program success story is the use of imatinib mesylate in chronic myelogenous leukemia (CML). Imatinib mesylate, a tyrosine kinase inhibitor, was granted AA following less than a 3-month review for CML in patients who were in blast crisis or had progressed to an advanced stage of the disease and who were no longer treatable with interferon. Imatinib mesylate demonstrated a 90.8% survival rate after 2 years of treatment. In 2003, the drug was granted full FDA approval status.18
Between December 1992 and December 2021 about half of AAs (n = 139) converted to traditional approvals. This took place in a median time of 3.2 years, thereby expediting the lengthy drug approval process.14
A review of nononcology drugs approved through the AA program between June 1992 and May 2018 found that 48 drugs for 57 nononcology indications were approved by the accelerated route and three-quarters of the indications (n = 43) were converted to regular approval after a median time of 53.1 months from AA.19
Harnessing the Power of Precision Medicine
The National Cancer Institute has established a new early clinical trial network called Experimental Therapeutics Clinical Trials Network (ET-CTN). The intent of ET-CTN is to identify the most appropriate therapy based on tumor type using biomarker-mining processes. These data could potentially result in "phenotype-to-genotype" profiles of treatment responders using whole exome sequencing.20
Might Offer Greater Gains in Quality-Adjusted Life-Years
A review of drugs approved through the FDA's expedited review process found greater gains in quality-adjusted life-years (QALYs) from pharmaceuticals approved through the AA process (n = 76 drugs) compared with the traditional approval process (n = 59 drugs) (0.370 vs. 0.031 QALYs, respectively).21
CONTROVERSY SURROUNDING THE AA PROGRAM
Do Surrogate Endpoints Represent Actual Clinical Benefit?
Questions have been raised about the applicability of surrogate endpoints used to gain AA status as to whether they correlate with their intended outcome. The strength of association of surrogates in oncology and clinically meaningful outcomes is often unknown, or worse yet, weak.22
A systematic review of trial-level meta-analyses examining the strength of association between surrogate endpoints and survival in oncology yielded concerning results. For more than one-half (52%), trial-level correlation between surrogate endpoint and clinical outcome was low, including the correlation between a surrogate endpoint and overall survival (OS). Studies incorporating surrogate endpoints often did not include information on the rates of surrogate improvement or improvement in OS. Most correlation of surrogacy appeared to be based on only a subset of clinical trials. Progression-free survival (PFS), which is the time from randomization to disease progression and is indicated by a >20% increase in the sum of diameters of target lesions with an absolute increase of at least 5 mm or any new lesion, or death, is often used as the primary endpoint of oncology trials and as the basis for regulatory approval. However, correlation with OS is low, especially in breast cancer, colorectal cancer, metastatic disease, gastric cancers, and non-small cell lung cancer. A case in point was the approval of bevacizumab for breast cancer based on PFS; improvement in PFS did not translate to an improvement in survival.23,24
Other oncologic agents approved for use in breast cancer based on PFS as the surrogate endpoint include everolimus and palbociclib. Liposomal doxorubicin was approved for the treatment of multiple myeloma based on a delay in time to progression when used in combination with bortezomib, but this surrogate endpoint was not validated for this type of malignancy.24
Another surrogate endpoint, pathologic complete response (pCR), which is often used in breast cancer trials, was found to correlate poorly with event-free survival and OS.24 pCR is the percentage of patients who achieve a pathologic complete response, which is defined as the absence of invasive neoplastic cells at microscopic examination of the primary tumor at surgery.23 Investigators have warned that it should not be used as the primary endpoint in regulatory neoadjuvant trials for early-stage breast cancer.25
Further, just because a surrogate endpoint is measurable does not mean that it is predictive of the desired outcome nor that it is meaningful. Overall, the association between surrogate endpoints and survival in oncology is poor.24
A review of 93 cancer drug indications granted AA by the FDA from December 11, 1992, through May 31, 2017, found that only about 20% (19 of 93) of confirmatory trials for oncology drugs approved by the AA process demonstrated improvement in OS. Concerningly, 37% of confirmatory trials used the same surrogate measures as for the preapproval trials instead of examining clinical outcomes as required.23
An investigation was conducted by the British Medical Journal in which manufacturers of 24 treatments that had been on the U.S. market >5 years were questioned as to their performance of phase IV confirmatory trials. Of the 253 drugs that received an AA from 1992 to 2020, 16 were withdrawn, 112 did not have confirmed clinical effectiveness trial data, and 24 have been on the U.S. market for 5 or more years. Despite being available on the market, two drugs were still in negotiation with the FDA over study trial design. An example uncovered by this investigation includes that midodrine has been on the market for 25 years but has not completed its confirmatory studies as of July 2021. However, as of 2009, the company had earned $257 million from the sale of this unproven drug.26
Even in the case of orphan drugs, there is need for improvement in the qualification process for biomarkers, as primary endpoints are often poorly defined in rare diseases.27,28 The National Organization for Rare Disorders commissioned a report that describes the history and opportunities for reform for the FDA's AA program from a rare disease perspective.29 A white paper has proposed a framework for assessing biomarker endpoints for rare and devastating diseases.30
In the case of the controversial AA approval of the antiamyloid monoclonal antibody aducanumab for Alzheimer's disease, an international group of professional and lay stakeholders questioned the biologic's approval based on surrogate makers because amyloid plaque does not correlate with clinical ratings, severity of disease, or progression. They stated that the FDA's position that amyloid plaque is "reasonably likely to predict" benefit is without merit.31
This concern was echoed in a commentary by a group of international researchers on the use of amyloid positron emission tomography (PET) as a single, primary surrogate efficacy measure in Alzheimer's disease immunotherapy trials. Among the reasons cited include that PET quantification of amyloid deposition is distorted and misrepresents the effect of investigational antiamyloid treatments due to a lack of specificity of the PET imaging probe; effects of amyloid-related imaging abnormalities; spillover high white matter signals, which are large, nonspecific amyloid PET signals in the white matter that diminish during immunotherapy; and questionable quantification models. The group called for the FDA to require "rigorous evidence of all imaging claims and irrefutable documentation that proposed treatments are clinically effective and harmless" and for repeat PET and MRI scans in Alzheimer's disease drug trials.32,33
There is a call to limit the use of surrogate endpoints to situations in which their utility has been demonstrated to robustly predict meaningful benefits or limited situations in which few treatment options are available. If used as a surrogate, it is essential that confirmatory studies be completed to determine the appropriateness of their continued use as a valid measure of the specified clinical outcome.22
Failure to Complete Confirmatory Studies in a Timely Manner
Since the association of surrogate endpoints to clinical outcome is a focus of debate, it is essential that confirmatory studies that include clinical outcomes be completed. Many drug approvals under the AA program have been based on small, statistically significant increases in surrogates that are of questionable reliability.22
A report from 2009 from the Government Accounting Office entitled "FDA Needs to Enhance Its Oversight of Drugs Approved on the Basis of Surrogate Endpoints" concluded that "weaknesses in FDA's monitoring and enforcement process hamper its ability to effectively oversee postmarketing studies. FDA has not routinely been reviewing sponsors' annual submissions on the status of studies in a timely manner. It has little in the way of readily accessible, comprehensive data to monitor studies' progression and does not consider such oversight a priority."34
The Office of Inspector General (OIG) conducted a review of the timeliness of confirmatory clinical trial completion for drugs granted an AA status. The OIG found for the 278 AAs granted between 1992 and December 31, 2021, 104 have incomplete confirmatory trials. Of those, 34% (35 of 104) have at least one trial past its original planned completion date, with four drug applications having confirmatory trials still pending more than 5 years to nearly 12 years past the original completion dates. The report also found that 13% of all AA drug applications have been withdrawn, with 50% of these withdrawals occurring since January 2021.35
The OIG also analyzed the cost to CMS/Medicare and Medicaid of drugs with incomplete confirmatory study data. They estimated that Medicare and Medicaid spent >$18 billion from 2018 to 2021 for 18 drugs that corresponded to the 35 drug applications granted AA with incomplete confirmatory trials past their original planned completion dates as of May 5, 2022.35
Increase in Boxed Warnings for AA Drugs
A review of nononcology drugs approved through the AA program between June 1992 and May 2018 found that the labeling of 47% of AA drugs was issued a boxed warning with a median time of 248.6 months from AA.19
In an evaluation of serious postmarketing safety signals within 2 years of FDA approval of new cancer drugs, only one of the 32 new molecular entities (NMEs) approved under the traditional pathway required a new warning and precaution, compared with seven of 23 new NMEs approved under the AA program. However, despite the increased incidence of substantial safety-related warning/precautions associated with AA drugs, the authors felt that these caveats did not significantly alter the benefit-risk profile of the drugs. On the other hand, ponatinib, which was approved via the AA program, was associated with significant safety concerns, necessitating the drug's temporary withdrawal because of the risk of developing arterial occlusive events. It also required the development of a Risk Evaluation and Mitigation Strategy program and a boxed warning.36
Use of Single-Arm Clinical Trials
Single-arm studies (SASs) are increasingly being used for AAs.37 Among the shortcomings of SASs are the inclusion of smaller, noncomparative safety datasets due the lack of a placebo-controlled group, inability to use time-to-event endpoints, limited generalizability to populations that are not included in the study, and inability to control for confounding factors.38,39 SASs used for AAs have been found to have a modest effect size and a high risk of bias.40
A review of oncology drug approvals published found that between January 1, 2002, and December 31, 2021, the FDA granted 176 drug/biologic approvals based on SASs, the majority (66%) of which were for AA status. About one-half of the SASs were used for approvals of NMEs or original biologics. SASs were conducted for 116 AAs. However, despite the less stringent study design, only 38% of AAs fulfilled their postmarketing requirement to verify the drug's clinical benefit. Further, 9% of drug approval withdrawals as of December 31, 2021, involved pharmaceuticals that were studied using the SAS design.39
Another study examining the use of SASs in oncology drugs granted AA status between 1992 to 2020 found that historical control data were often not clearly reported. Investigators called for greater transparency in historical control analysis. Researchers found that 47% of the 254 AAs during this study period utilized SASs. The most common oncological indications for which SASs were conducted included for urothelial cancer, multiple myeloma, and thyroid cancer. Although 53% of the AA indications converted to traditional approval, five indications were withdrawn from the market.37
Given its concerns with the use of SASs, in March 2023 the FDA issued a draft guidance for industry on clinical trial considerations to support the AA of oncology therapeutics in which it recommended the randomized, controlled trial as the preferred approach to support an application for AA status and where it identified specific design, efficacy, and trial analysis considerations in the limited circumstances where the performance of an SAS may be warranted (www.fda.gov/regulatory-information/search-fda-guidance-documents/clinical-trial-considerations-support-accelerated-approval-oncology-therapeutics).
Withdrawal of AA Status
One of the major concerns expressed has been the difficulty in withdrawing AAs of medications for which confirmatory studies have not shown that the drug is beneficial (and/or may be potentially harmful) or if the confirmatory studies have not been completed. This has resulted in undue delays in withdrawing drugs whose benefits have not been confirmed in clinical trials. (See TABLE 1.)
In a study of nononcology drugs approved through the AA program between June 1992 and May 2018, 20% of the 86 drugs had not fulfilled their postapproval confirmatory trial requirements. Of those that did, the median time to confirmatory trial completion was 39.4 months. Although 10% of confirmatory studies failed to verify clinical efficacy, only 2% of indications were withdrawn. These drugs remained on the market for 136 months after AA.
Mafenide acetate 5% topical solution (Sulfamylon) was approved on June 5, 1998, and was indicated as an adjunctive topical antimicrobial agent to control bacterial infection when used under moist dressings over meshed autograft on excised burn wounds. It took nearly 24.5 years to withdraw the AA of this drug. By November 22, 2022, Mylan, the originator company, had not completed the required confirmatory studies. The company indicated that conducting the required confirmatory studies was not feasible. Sulfamylon's drug approval status was subsequently withdrawn.41
Listed below are other drugs/indications for which confirmatory studies have failed to demonstrate benefit and their time to withdrawal:
- Synercid (dalfopristin/quinupristin): vancomycin-resistant Enterococcus faecium (11 years, 1 month)
- Ethyol (amifostine): reduce cumulative renal toxicity from cisplatin in non-small-cell lung cancer (10 years)
- Blenrep (belantamab mafodotin-blmf): relapsed or refractory multiple myeloma (2 years, 6 months)
- Keytruda (pembrolizumab): recurrent or locally advanced or metastatic gastric or gastric junction adenocarcinoma whose tumors express PD-L1 (4 years, 4 months)
- ecentriq (atezolizumab): unresectable locally advanced or metastatic triple-negative breast cancers that express PD-L1 (2 years, 6 months), locally advanced or metastatic urothelial carcinoma not eligible for cisplatin and whose tumor expresses PD-L1 (5 years, 7 months), and locally advanced or metastatic urothelial carcinoma (4 years, 10 months)
- Opdivo (nivolumab): metastatic small-cell lung cancer (2 years, 4 months)
- Imfinzi (durvalumab): locally advanced or metastatic urothelial carcinoma (3 years, 9 months)
- Imbruvica (ibrutinib): marginal zone lymphoma (6 years, 4 months) and mantle cell lymphoma (9 years, 6 months)
- Lartruvo (olaratumab): soft tissue sarcoma that is not curable with radiation or surgery (3 years, 4 months)
- Istodax (romidepsin): peripheral T-cell lymphoma (10 years, 1 month)
- Avastin (bevacizumab): metastatic HER2-negative breast cancer (3 years, 8 months)
- Iressa (gefitinib): locally advanced or metastatic non-small-cell lung cancer (8 years, 11 months).
Other drugs/indications for which confirmatory studies were not done or completed and their time to withdrawal include9:
- Luveris (lutropin alfa): stimulation of follicular development in infertile hypogonadotropic hypogonadal women with profound LH deficiency (11 years, 6 months)
- Gavreto (pralsetinib): advanced or metastatic RET-mutant medullary thyroid cancer (2 years, 7 months)
- Copiktra (duvelisib): relapsed or refractory follicular lymphoma (3 years, 2 months)
- Farydak (panobinostat): multiple myeloma (7 years, 1 month)
- Zydelig (idelalisib): relapsed follicular B-cell non-Hodgkin's lymphoma and relapsed small lymphocytic lymphoma (7 years, 6 months)
- Marqibo (vincristine sulfate liposomal): Philadelphia chromosome-negative acute lymphocytic leukemia in second relapse or greater (9 years, 8 months)
- Bexxar (tositumomab and iodine 1-131 tositumomab): relapsed or refractory low-grade follicular or transformed CD20+ non-Hodgkin's lymphoma not treated with rituximab (8 years, 10 months)
- Celebrex (celecoxib): adjunctive therapy to reduce the number of adenomatous colorectal polyps in patients with familial adenomatous polyposis (12 years, 5 months).
Lack of Available Information Regarding a Drug's AA Status
A retrospective analysis was conducted of label information for drugs approved under the AA process from January 1, 1992, to December 31, 2020. Researchers found that 13% of prescribing information labeling lacked sufficient information that the drug's approval either was via the AA process or was based on surrogate endpoints, not clinical outcome measures. The investigators found that 7% of product labels did not mention that the drug had been approved via the accelerated route, although they did describe the use of surrogate markers. Another 4% of labels did not mention either the use of the AA process or of surrogate endpoints. The last 2% mentioned that the drug was approved via the AA process, but there was no mention of the use of surrogate endpoints. None of the labels for any of the 110 AA indications across 62 drugs mentioned or described the clinical outcome being evaluated in the confirmatory studies.42 This demonstrates that clinicians are prescribing these medications and are engaged in the clinical decision-making process without being fully informed that these drugs were approved based on surrogate, not clinical, outcomes.
CALL FOR LEGISLATIVE REFORM TO THE AA PROGRAM
In April 2021, the Institute for Clinical and Economic Review released a white paper that proposed recommendations for strengthening the AA process. The report provides an analysis of potential policy reforms and their impact on uncertainty, access, innovation, and costs.18
The AA Integrity Act (AAIA, H.R. 6963), which was introduced in the House of Representatives on March 7, 2022, by Representative Frank Pallone Jr. (D-NJ), calls for the establishment of requirements relating to postapproval studies for drugs that receive AA for serious or life-threatening diseases or conditions, such as requiring product sponsors to agree in advance to conduct such studies and terminating product approval if certain benchmarks are not met.
More specifically, the bill requires the completion of appropriate, adequate, and well-controlled postapproval studies to verify and describe the predicted effect on irreversible morbidity or mortality or other clinical benefits. The Act would require that confirmatory trials are initiated prior to AAIA and include information on enrollment targets, study protocol and milestones, and the target date of study completion. The federal government would be allowed to refuse to approve a product under the AA program until confirmatory studies are underway by the sponsor.43
The federal government would also be given broader authority to withdraw approval of an AA if the sponsor fails to conduct any required postapproval studies; achieve agreed-upon enrollment targets, milestones, or timely study completion goals; submit reports; verify or demonstrate a beneficial effect; show that the drug is safe or effective for the conditions of use; or if the sponsor disseminates false or misleading promotional materials about the AA product.43
The bill also advocates for the automatic expiration 1 year after the passage of any target date of study completion included in an agreement. In no case should the study completion date exceed 5 years after the date from which the product is approved, unless the predicted effect on irreversible morbidity or mortality or other clinical benefit of the product has been verified or the federal government has determined that adequate progress has been made on completion of required postapproval studies.43
For clinicians to be able to identify that a drug has been approved through the AA process (not the traditional approval route), the act calls for a statement to be included in the labeling that the product was approved under AA and that the continued approval of the product is contingent upon the completion of postmarketing studies to verify clinical benefit. The product labeling should also identify the clinical endpoint that is under study and the limitations, if known, of the surrogate or immediate endpoint to determine the anticipated outcome. This information should provide a succinct description of the product and should indicate any uncertainty about anticipated clinical benefit as well as a discussion of available evidence on the drug's proposed clinical benefit.43
The drug sponsor should provide an update on the study progress based on the data points agreed on such as targets, milestones, and other required information upon enrollment. This information should be submitted no later than 90 days after the approval of the drug and every 90 days thereafter, until the study is completed or terminated.43
CONCLUSION
Although the intent of the FDA's AA is to increase access to potentially life-saving medications for serious or life-threatening diseases, it has also allowed for the approval of medications based on surrogate endpoints, which may not adequately correlate with the clinical outcome that they are intended to predict. Confirmatory studies for pharmaceuticals approved via the AA pathway need to be conducted in a timely manner. These confirmatory studies should not involve continued study of surrogate endpoint data. Rather, these studies need to determine if the surrogate endpoint is truly reflective of clinical outcomes. When a lack of benefit is demonstrated, these drugs should be quickly removed from the market. Further, the FDA needs to remove drugs or biologicals from the market when confirmatory testing has not been initiated or completed as planned.
Pharmacists need to be aware of the strength and weakness of the data available supporting the drugs and biologicals that have been approved via the AA process, which is less vigorous than what is obtained from the traditional drug-approval process. Pharmacists need to be proactive in monitoring patients for both safety and efficacy if they are receiving pharmaceuticals approved via the AA pathway.
The content contained in this article is for informational purposes only. The content is not intended to be a substitute for professional advice. Reliance on any information provided in this article is solely at your own risk.
REFERENCES
1. FDA. Fast track, breakthrough therapy, accelerated approval, priority review. www.fda.gov/patients/learn-about-drug-and-device-approvals/fast-track-breakthrough-therapy-accelerated-approval-priority-review. Accessed July 15, 2023.
2. FDA. Accelerated approval. www.fda.gov/patients/fast-track-breakthrough-therapy-accelerated-approval-priority-review/accelerated-approval. Accessed July 15, 2023.
3. FDA. Surrogate endpoint resources for drug and biologic development. www.fda.gov/drugs/development-resources/surrogate-endpoint-resources-drug-and-biologic-development. Accessed July 15, 2023.
4. FDA. New drug approvals 2022–Advancing health through innovation. www.fda.gov/media/164429/download?attachment. Accessed July 15, 2023.
5. Sachs RE, Gavulic KA, Donohue JM, et al. Recent trends in Medicaid spending and use of drugs with U.S. Food and Drug Administration Accelerated Approval. JAMA Health ForumHealth Forum. 2021;2(10):e213177.
6. Federal Food, Drug, and Cosmetic Act [As Amended Through P.L.117–328, Enacted December 29, 2022]. As Amended Through P.L. 117-328, Enacted December 29, 2022/ Published May 19, 2023. www.govinfo.gov/content/pkg/COMPS-973/pdf/COMPS-973.pdf. Accessed July 15, 2023.
7. FDA. 21st Century cures: announcing the establishment of a surrogate endpoint table; establishment of a public docket; request for comments. Fed Reg. 2018;83(210):54593-54594.
8. FDA. Drug development tool process under the 21st Century Cures Act and Prescription Drug User Fee Act VI; Public Meeting; Request for Comments. Fed Reg. 2018;83(219):56347-56349.
9. FDA. Accelerated approval program. www.fda.gov/drugs/nda-and-bla-approvals/accelerated-approval-program. Accessed August 14, 2023.
10. Sutter S. The Pink Sheet. Two years in US accelerated approval withdrawals. January 25, 2023. https://pink.pharmaintelligence.informa.com/PS147526/Two-Years-In-US-Accelerated-Approval-Withdrawals. Accessed July 15, 2023.
11. Kepplinger EE. FDA's expedited approval mechanisms for new drug products. Biotechnol Law Rep. 2015;34(1):15-37.
12. Drugs.com. FDA drug approval process. www.drugs.com/fda-approval-process.html. Accessed July 15, 2023.
13. FDA. Project Confirm. www.fda.gov/about-fda/oncology-center-excellence/project-confirm. Accessed July 15, 2023.
14. Beakes-Read G, Neisser M, Frey P, et al. Analysis of FDA's Accelerated Approval Program performance December 1992–December 2021. Ther Innov Regul Sci. 2022;56(5):698-703.
15. FDA. Table of surrogate endpoints that were the basis of drug approval or licensure. www.fda.gov/drugs/development-resources/table-surrogate-endpoints-were-basis-drug-approval-or-licensure. Accessed July 15, 2023.
16. Dunoyer M. Accelerating access to treatments for rare diseases. Nat Rev Drug Discov. 2011;10(7):475-476.
17. Monge AN, Sigelman DW, Temple RJ, et al. Use of US Food and Drug Administration expedited drug development and review programs by orphan and nonorphan novel drugs approved from 2008 to 2021. JAMA Netw Open. 2022;5(11):e2239336.
18. Kaltenboeck A, Mehlman A, Pearson SD. Institute for Clinical and Economic Review. Strengthening the Accelerated Approval pathway: an analysis of potential policy reforms and their impact on uncertainty, access, innovation, and costs. April 26, 2021. https://icer.org/wp-content/uploads/2021/04/Strengthening-the-Accelerated-Approval-Pathway-_-ICER-White-Paper-_-April-2021.pdf. Accessed July 15, 2023.
19. Omae K, Onishi A, Sahker E, et al. US Food and Drug Administration Accelerated Approval Program for nononcology drug indications between 1992 and 2018. JAMA Netw Open. 2022;5(9):e2230973.
20. Bando H, Takebe N. Recent innovations in the USA National Cancer Institute-sponsored investigator initiated Phase I and II anticancer drug development. Jpn J Clin Oncol. 2015;45(11):1001-1006.
21. Chambers JD, Thorat T, Wilkinson CL, et al. Drugs cleared through the FDA's expedited review offer greater gains than drugs approved by conventional process. Health Aff (Millwood). 2017;36(8):1408-1415.
22. Kemp R, Prasad V. Surrogate endpoints in oncology: when are they acceptable for regulatory and clinical decisions, and are they currently overused? BMC Med. 2017;15(1):134.
23. Gyawali B, Hey SP, Kesselheim AS. Assessment of the clinical benefit of cancer drugs receiving accelerated approval. JAMA Intern Med.2019;179(7):906-913.
24. Prasad V, Kim C, Burotto M, et al. The strength of association between surrogate end points and survival in oncology: a systematic review of trial-level meta-analyses. JAMA Intern Med.2015;175(8):1389-1398.
25. Conforti F, Pala L, Sala I, et al. Evaluation of pathological complete response as surrogate endpoint in neoadjuvant randomised clinical trials of early stage breast cancer: systematic review and meta-analysis. BMJ. 2021;375:e066381.
26. Mahase E. FDA allows drugs without proven clinical benefit to languish for years on accelerated pathway. BMJ. 2021;374:n1898.
27. Kakkis ED, Kowalcyk S, Bronstein MG. Accessing the accelerated approval pathway for rare disease therapeutics. Nat Biotechnol. 2016;34(4):380-383.
28. Miyamoto BE, Kakkis ED. The potential investment impact of improved access to accelerated approval on the development of treatments for low prevalence rare diseases. Orphanet J Rare Dis. 2011;6:49.
29. National Organization for Rare Disorders. FDA’s Accelerated Approval pathway: a rare disease perspective. https://rarediseases.org/wp-content/uploads/2021/06/NRD-2182-Policy-Report_Accelerated-Approval_FNL.pdf. Accessed August 26, 2023.
30. Kakkis ED, O'Donovan M, Cox G, et al. Recommendations for the development of rare disease drugs using the accelerated approval pathway and for qualifying biomarkers as primary endpoints. Orphanet J Rare Dis. 2015;10:16.
31. Whitehouse P, Gandy S, Saini V, et al. Making the case for accelerated withdrawal of aducanumab. J Alzheimers Dis. 2022;87(3):1003-1007.
32. Høilund-Carlsen PF, Revheim ME, Alavi A, et al. Amyloid PET: a questionable single primary surrogate efficacy measure on Alzheimer immunotherapy trials. J Alzheimers Dis. 2022;90(4):1395-1399.
33. Høilund-Carlsen PF, Revheim ME, Alavi A, et al. FDG PET (and MRI) for monitoring immunotherapy in Alzheimer disease. Clin Nucl Med. 2023;48(8):689-691.
34. Government Accounting Office. FDA needs to enhance its oversight of drugs approved on the basis of surrogate endpoints. September 23, 2009. www.gao.gov/assets/gao-09-866.pdf. Accessed July 15, 2023.
35. Office of Inspector General. Delays in confirmatory trials for drug applications granted FDA’s Accelerated Approval raise concerns. https://ig.hhs.gov/oei/reports/OEI-01-21-00401.asp. Accessed July 15, 2023.
36. Kim J, Nair A, Keegan P, et al. Evaluation of serious postmarket safety signals within 2 Years of FDA approval for new cancer drugs. Oncologist. 2020;25(4):348-354.
37. Ribeiro TB, Bennett CL, Colunga-Lozano LE, et al. Increasing FDA-accelerated approval of single-arm trials in oncology (1992 to 2020). J Clin Epidemiol. 2023;159:151-158.
38. Zheng L, Rosenkranz SL, Taiwo B, et al. The design of single-arm clinical trials of combination antiretroviral regimens for treatment-naive HIV-infected patients. AIDS Res Hum Retroviruses. 2013;29(4):652-657.
39. Agrawal S, Arora S, Amiri-Kordestani L, et al. Use of single-arm trials for US Food and Drug Administration drug approval in oncology, 2002-2021. JAMA Oncol. 2023;9(2):266-272.
40. Ribeiro TB, Colunga-Lozano LE, Araujo APV, et al. Single-arm clinical trials that supported FDA accelerated approvals have modest effect sizes and were at high risk of bias. J Clin Epidemiol. 2022;148:193-195.
41. FDA. Mylan Institutional, Inc. Withdrawal of approval of a New Drug Application for Sulfamylon® (Mafenide Acetate, USP) Powder for 5% Topical Solution. Fed Reg. 2022;87(229):73561.
42. Ballreich J, Socal M, Bennett CL, et al. Accelerated approval drug labels often lack information for clinical decision-making. Pharmacotherapy. 2023;43(4):300-304.
43. H.R.6963–Accelerated Approval Integrity Act of 2022. 117th Congress (2021-2022). www.congress.gov/bill/117th-congress/house-bill/6963/text. Accessed July 15, 2023.