The Pharmacist’s Role in Testicular Cancer Treatment
RELEASE DATE
June 30, 2020
EXPIRATION DATE
June 30, 2022
FACULTY
Ashley Barlow, PharmD
PGY1 Pharmacy Resident
University of Maryland Medical Center
Baltimore, Maryland
Brooke Barlow, PharmD
PGY1 Pharmacy Resident
University of Kentucky Health Care
Lexington, Kentucky
Ali McBride, PharmD, MS, BCOP, FASHP, FAzPA
Clinical Coordinator of Hematology/Oncology
The University of Arizona Cancer Center
Tucson, Arizona
FACULTY DISCLOSURE STATEMENTS
Drs. Barlow, Barlow, and McBride have no actual or potential conflicts of interest in relation to this activity.
Postgraduate Healthcare Education, LLC does not view the existence of relationships as an implication of bias or that the value of the material is decreased. The content of the activity was planned to be balanced, objective, and scientifically rigorous. Occasionally, authors may express opinions that represent their own viewpoint. Conclusions drawn by participants should be derived from objective analysis of scientific data.
ACCREDITATION STATEMENT
 Pharmacy
Pharmacy
Postgraduate Healthcare Education, LLC is accredited by the Accreditation Council for Pharmacy Education as a provider of continuing pharmacy education.
UAN: 0430-0000-20-057-H01-P
Credits: 2.0 hours (0.20 ceu)
Type of Activity: Knowledge
TARGET AUDIENCE
This accredited activity is targeted to pharmacists. Estimated time to complete this activity is 120 minutes.
Exam processing and other inquiries to:
CE Customer Service: (800) 825-4696 or cecustomerservice@powerpak.com
DISCLAIMER:
Participants have an implied responsibility to use the newly acquired information to enhance patient outcomes and their own professional development. The information presented in this activity is not meant to serve as a guideline for patient management. Any procedures, medications, or other courses of diagnosis or treatment discussed or suggested in this activity should not be used by clinicians without evaluation of their patients’ conditions and possible contraindications or dangers in use, review of any applicable manufacturer’s product information, and comparison with recommendations of other authorities.
GOAL
To supply pharmacists with in-depth information about testicular cancer, including epidemiology, risk factors, treatment, and management strategies for chemotherapy toxicities.
OBJECTIVES
After completing this activity, the pharmacist should be able to:
- Describe the epidemiology of and risk factors for testicular germ-cell tumors (tGCT).
- Identify the surgical options and chemotherapy regimens for tGCT management based on seminoma and nonseminoma subtypes.
- List the medication-related toxicities associated with chemotherapy regimens and measures to improve outcomes in patients undergoing tGCT treatment.
- Discuss the role of fertility preservation in patients undergoing surgery or chemotherapy for tGCT.
ABSTRACT: Testicular cancer is one of the most curable solid tumors, with an estimated 10-year survival rate of about 95%. Testicular germ-cell tumors comprise 95% of all testicular cancers and are classified into two histologic subtypes: seminoma and nonseminoma. Regardless of the histologic subtype, cisplatin-based combination chemotherapy remains the cornerstone treatment, with adjunctive surgical or radiotherapy interventions considered for patients based on tumor burden. Given the remarkable long-term survival rates in patients with testicular cancer, pharmacists serve a vital role in helping patients weigh the risks and benefits of chemotherapy options, in addition to monitoring and managing its immediate and long-term toxic effects to maximize quality of life after therapy.
Testicular cancer is rare, accounting for fewer than 1% of all cancer cases in males and occurring in one of every 250 males.1 The incidence has steadily risen over the past few decades, with 9,610 new cases diagnosed in 2019.2 This form of cancer affects mainly young and middle-aged men, and the average age of onset is 33 years. Historically, the outcome for patients with testicular cancer has been dismal, with a 5-year survival rate of less than 30%; however, recent advances in early detection and management have markedly improved the 10-year survival rate to approximately 95%, rendering it one of the most curable solid tumors.1 With early diagnosis and tumor resection or commencement of chemotherapy with or without radiotherapy (RT), patients with early-stage disease have a 15-year survival rate exceeding 99%.3 Even patients who present with advanced disease have a good prognosis and an overall survival rate of 70% to 80%.4
The World Health Organization stratifies testicular cancer into distinct subtypes: testicular germ-cell tumors (tGCT) and testicular non–germ-cell tumors (t-nGCT). The tGCT subtype is further subdivided into seminoma and nonseminoma. The subtype classification is based on histologic composition, the germ cell involved, and the embryonic lineages present.5 The most common subtype of testicular cancer, comprising 95% of all cases, is tGCT.2 Given the rarity of t-nGCT, this article will focus specifically on tGCT.
Pathophysiology
The pathogenesis of tGCT involves abnormal alterations in the developmental process of the germ cell toward full spermatogenesis. Each germinal cell and layer of embryonic tissue are affected at different phases of maturation, leading to significant heterogeneity in disease histology. Seminomas originate in the seminiferous tubules and consist of syncytial trophoblastic cells that express beta–human chorionic gonadotropin (beta-hCG) on the cell surface. Seminomas constitute a majority of tGCT and follow a less aggressive disease course than nonseminomas. Nonseminomas develop from multiple embryonic cell subtypes.6 Nonseminomas express heterogeneous tumor markers such as alpha-fetoprotein (AFP), beta-hCG, and lactate dehydrogenase (LDH), and they are more likely than seminomas to progress to metastatic disease.6
Risk Factors
Unfortunately, most risk factors for tGCT are non-modifiable, with genetic (Klinefelter’s syndrome) and inherited conditions having the greatest influence.2 The risk increases 8-fold to 10-fold in men with 1st-degree or 2nd-degree relatives who have a history of tGCT.7 The greatest incidence of tGCT is in postpubertal males, with a slight variance between subtypes. Seminomas most often occur in the 2nd or 3rd decade of life, compared with the 4th decade of life for nonseminomas. Cryptorchidism, a condition in which one or both testes fail to descend into the scrotum at birth, confers a fivefold increase in risk over persons with normal testicular development.8 Caucasians have a greater predisposition toward developing tGCT; however, African Americans tend to have more aggressive disease.7 HIV is a risk factor specific to seminoma tGCT development.1,9 Within 15 years of initial diagnosis of a unilateral tumor or testicular intraepithelial neoplasia, approximately 2% of patients will develop tGCT in the contralateral testis.2 One modifiable risk factor for tGCT that has been observed in epidemiologic data is frequent, long-term marijuana use.10
Clinical Presentation
Most tGCT follow an indolent course and may go unrecognized for months to years before clinical symptoms develop. The most common presenting feature is a unilateral mass or hardened nodule in the scrotum. This may be accompanied by diffuse testicular pain and swelling that closely mimics nonmalignant conditions such as orchitis and epididymitis.2 Without vigilant examination and heightened awareness of the potential overlap with other conditions, the diagnosis of tGCT may be delayed. Nonlocalized symptoms in the abdomen or chest can develop if the tumor invades extragonadal regions, most commonly the retroperitoneum and the mediastinum. Rarely, patients present with enlarged lymph nodes of the lower neck or upper chest, a retroperitoneal mass, cachexia, gynecomastia, or venous thromboembolisms. The presence of these symptoms is suggestive of more advanced metastatic disease.6
Diagnostic Evaluation
A comprehensive diagnostic assessment is essential to determine potential risk factors and evaluate the disease stage. Diagnostic assessment starts with taking a comprehensive history and performing physical examination, followed by a chest x-ray and chemistry profile to obtain serum tumor markers. When a unilateral testicular mass is present on physical examination, transscrotal ultrasound is performed to further assess its extent and etiology. If the ultrasound confirms the presence of a malignant mass, orchiectomy (surgery to remove one or both diseased testes and the full spermatic cord) may be performed.2,11 This serves as a diagnostic and therapeutic intervention, as the full tumor can be removed and the resected tissue used to determine the histologic sub-type in order to guide treatment. Unlike other cancers, in which biopsy is the gold standard for diagnosis, biopsy is relatively contraindicated in tGCT because it can increase the risk of local or atypical regional recurrence.2 Another critical test is computed tomography (CT) of the abdomen, pelvis, and chest to detect any alternative disease sites. If signs and symptoms of advanced or metastatic disease are present, along with elevated tumor markers, magnetic resonance imaging of the brain and bone should be performed to rule out metastasis.6,11
Staging and Tumor Markers
Tumor staging for tGCT is performed using the American Joint Committee on Cancer (AJCC) tumor, node, metastasis (TNM) staging criteria (TABLE 1).12 Tumors are characterized as clinical stage I (disease confined to the testes and epididymis), II (lymph node involvement), or III (metastasis to visceral organs). For stage III, the International Germ Cell Cancer Collaborative Group’s risk classification is used instead of the TNM criteria, involving the categories of good, intermediate, and poor risk.13 Unique to the staging of testicular cancer is the incorporation of the S stage, which assesses the level of serum tumor markers.12 Serum tumor markers play a fundamental role in determining the stage of disease after orchiectomy and provide valuable information about the potential for metastasis, disease prognosis, and recurrence risk postoperatively.4,6,11 It is vital to use serum tumor markers to differentiate between seminoma and nonseminoma because their treatments differ.
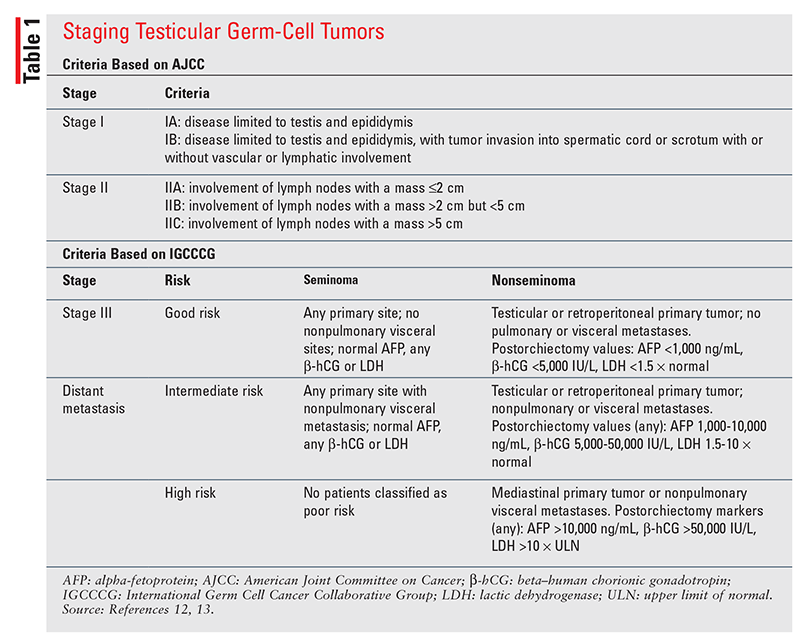
Serum tumor markers are an added element to the staging criteria for tGCT. Serum AFP is a marker specific to the diagnosis of nonseminomas. If after orchiectomy the histology is suspected to be seminoma, elevations in serum AFP would indicate a mixed histology with nonseminoma.2 Beta-hCG is expressed on the surface of seminoma cell types; therefore, trends in beta-hCG serve as a marker of disease control or progression after treatment.4,11 LDH is a nonspecific tumor marker and is used mainly to help determine tumor burden, elucidate rate of cellular growth, and predict prognosis.2,12 Tumor markers may also be used to determine response to chemotherapy. The terminal half-lives of AFP (5 days) and beta-hCG (~36 hours) should be considered, and slower- than-expected reductions may indicate poor prognosis, treatment resistance, or disease progression.14 However, these tumor markers are nonspecific and may suggest the presence of other malignancies (e.g., bladder cancer, hepatocellular cancer, adenocarcinomas) or comorbid conditions (hypogonadism, marijuana use, hyperthyroidism).2
Treatment
The National Comprehensive Cancer Network (NCCN) guidelines for tGCT base treatment recommendations on the disease’s clinical stage and histologic subtype.11 As mentioned previously, orchiectomy is performed for diagnosis in most cases. Following orchiectomy, treatment centers around reducing the risk of recurrence, optimizing quality of life, and minimizing the risk of treatment-related adverse effects.11 Cytotoxic chemotherapy plus nerve-sparing retroperitoneal lymph node dissection (RPLND) for nonseminomas and RT for seminomas are the two primary adjunctive treatment modalities. No specific driver mutation has been identified to offer targeted therapy for tGCT. However, targeted therapy may not be a moving target for treatment given that tGCTs are exquisitely sensitive to cytotoxic chemotherapy.6 Initiation of chemotherapy prior to diagnosis or surgery is usually not warranted, although it may be considered for patients with markedly elevated AFP, retroperitoneal or mediastinal tGCT, or significant symptoms and a high disease burden.11
Stage I
For seminomas and nonseminomas, stage I disease can be managed with orchiectomy alone. Post orchiectomy, adjunctive therapies may be offered based on the risk of relapse. Because overall survival approaches 100% for stage I disease, chemotherapy and RT are often deferred owing to the higher risk of long-term side effects (i.e., secondary malignancies) versus overall benefit.2 The NCCN guidelines strongly prefer surveillance post orchiectomy for pure stage I seminomas and nonseminomas without high-risk features.11 In a patient with seminoma who is deemed to be at risk for relapse or nonseminoma with high-risk features such as lymphovascular invasion (LVI), spermatic-cord infiltration, and scrotal invasion, the following postorchiectomy treatment options may be considered.
Seminoma: For stage I seminoma managed with surveillance alone, there is a 13% to 20% chance of relapse at 5 years without adjuvant treatment.15 Higher rates of relapse are observed in patients with tumor invasion of the rete testis, patients with a large tumor burden (4 cm or greater), and those unable to adhere to postorchiectomy surveillance visits.12 Two accepted strategies exist for reducing the risk of relapse: 1) one or two cycles of carboplatin targeting an AUC of 7, and 2) RT. The decision on whether to pursue one or two cycles of carboplatin should be based on patient-specific factors and risk of adverse effects. A study comparing one cycle of carboplatin, two cycles of carboplatin, and active surveillance found that disease-free survival was 100% in each group, although crude relapse rates were higher in patients receiving one (5%) versus two carboplatin cycles (1.5%), and relapse rates for both options were superior to those for surveillance (8.2%).16 A majority of patients who failed one cycle had a large tumor; therefore, two cycles may be considered in this setting.
Nonseminoma: Patients with nonseminomas and high-risk features, particularly LVI, have a 50% risk of metastasis without adjunctive treatment.2 If LVI is absent, there is a 14% to 22% risk of relapse, and determining adjunctive therapy in this population is based on patient-specific risks.17 The primary recommended treatment for high-risk patients includes adjuvant chemotherapy with one cycle of bleomycin, etoposide, and cisplatin (BEP) or nerve-sparing RPLND.11 In nerve-sparing RPLND, the lymph nodes are dissected via a particular technique that spares removal of or interference with the testicular nerves.6 This strategy has improved rates of postoperative fertility, minimizes the risk of postoperative retrograde ejaculation, and reduces the retroperitoneal recurrence rate to less than 5%.11 Adjunctive therapy with BEP is recommended over surveillance based on a prospective trial in which relapse rates at 5 years decreased significantly to 3.2% in patients with LVI, with an overall survival rate of 100%.18 Investigators have also assessed the efficacy of two cycles to further reduce relapse rates. However, this strategy is not recommended owing to the increased risk of hearing damage and loss, cardiovascular (CV) conditions, hypertension, and neuropathy from cisplatin without any benefit in overall survival.19
Stage IS
This stage is characterized by stage I disease with persistent elevation of serum tumor markers post orchiectomy. Serum tumor markers that remain elevated 2 to 3 weeks after surgery should prompt additional imaging for metastatic disease beyond the retroperitoneum.
Stage IS disease is rare for seminomas and may indicate mixed histology with nonseminomas or metastasis. If mixed histology is found, the treatment course should follow that for nonseminomas to address the aggressive disease course of nonseminomas. For nonseminomas, a regimen of either three cycles of BEP or four cycles of etoposide and cisplatin (EP) is recommended. Both regimens are category 1 recommendations, and either regimen is preferable to initial RPLND because stage IS patients nearly always have disseminated disease when there is evidence of persistently elevated biomarkers.11
Stage II
Seminoma: Primary treatment of stage IIA and IIB seminomas includes RT or chemotherapy with three cycles of BEP or four cycles of EP.11 Retrospective studies reveal a lower response to RT in stage IIB disease with bulky tumor size (3 cm or greater); therefore, the NCCN guidelines recommend chemotherapy. In stage IIC disease, treatment is similar to that for stage III disease (discussed below).11
Nonseminoma: All patients with stage IIA disease undergo nerve-sparing RPLND, which serves to remove the tumor within the lymphatic system. Following RPLND, either surveillance or chemotherapy is offered, depending on the number and size of lymph nodes involved and the presence or absence of serum tumor markers. In many cases of stage IIA disease with no regional lymph node metastasis, primary treatment with nerve-sparing RPLND followed by surveillance is curative.11 When the tumor has invaded up to five lymph nodes (pN1), adjunctive chemotherapy with two cycles of EP may be considered to prevent invasion into adjacent lymph nodes.11 When lymphatic involvement exceeds five nodes or the disease is more aggressive (stage IIB/C), more than 50% of cases will relapse in the absence of adjunctive chemotherapy.11 Cisplatin-based chemotherapy has been a cornerstone treatment demonstrating remarkable improvements in survival and reductions in relapse to less than 1%.2 Two cycles of adjunctive chemotherapy with EP are preferred for pN2 disease, in which the tumor affects more than five lymph nodes but is less than 5 cm. Three or four cycles of BEP or four cycles of EP are recommended for pN3 disease in which lymph-node involvement exceeds 5 cm. For stage IIB/C disease, chemotherapy and nerve-sparing RPLND have comparable efficacy, and they improve relapse-free survival to approximately 98%.2
Stage III
For seminomas and nonseminomas, stage III disease involves tumor metastasis and serum biomarker elevation. Based on the location and extent of disease and biomarker elevation after orchiectomy, the disease is classified as good-risk, intermediate-risk, or poor-risk.11
Seminoma: Given the improved survival rates and less aggressive course of seminomas, classification is limited to good or intermediate risk. Stage IIC/III seminomas are considered good-risk, except for stage IIIC, which is deemed intermediate-risk based on the presence of nonpulmonary visceral metastases (e.g., bone, liver, brain). Chemotherapy is recommended as first-line treatment post orchiectomy for all stage IIC/III patients.
In patients with good-risk disease, three cycles of BEP or four cycles of EP are recommended. More intensive chemotherapy is required for intermediate-risk disease, with the preferred regimen being four cycles of BEP; four cycles of etoposide, mesna, ifosfamide, and cisplatin (VIP) are an alternative. VIP is reserved for patients with a contraindication to bleomycin because, compared with BEP, it does not improve efficacy and has remarkably higher rates of toxicities, specifically pulmonary toxicities.11
Nonseminoma: Good-risk disease, also referred to as stage IIIA, has a cure rate of 90% when treated with three cycles of BEP or four cycles of EP.4,20 For intermediate-risk disease, treatment with four cycles of BEP (preferred) or four cycles of VIP is recommended, as it has an overall cure rate of approximately 70%.11 Poor-risk disease is managed with four cycles of BEP, and VIP is a second-line alternative for patients who may not tolerate bleomycin. Unfortunately, despite chemotherapy, these patients fare poorly; fewer than 50% respond to BEP, and there is a 30% risk of death from disease progression.21
Relapsed/Refractory Disease
Despite high initial treatment responses to front-line chemotherapy, it is estimated that approximately 20% of patients will relapse, necessitating initiation of salvage therapy.1 It is recommended that these patients be transferred to a high-volume center for further management by experts in this rare tumor type. Surgical resection alone may be considered for patients with early recurrence (<2 years) if the tumor is locally confined.2,11 For patients with late recurrence or disseminated disease, three management strategies exist: conventional-dose chemotherapy (CD-cT) followed by autologous stem-cell transplantation (ASCT); high-dose salvage chemotherapy (HD-cT) followed by ASCT; and enrollment in a clinical trial.2,11 CD-cT regimens include either paclitaxel, ifosfamide, and cisplatin or vinblastine, ifosfamide, and cisplatin. HD-cT regimens include high-dose carboplatin-etoposide or paclitaxel plus ifosfamide followed by high-dose carboplatin and etoposide.11 These preconditioning regimens consist of broad-spectrum, highly toxic chemotherapy that removes residual tumor. Owing to significant myelosuppression, the bone marrow is rescued with an ASCT.
Unfortunately, data are lacking regarding the superiority of CD-cT or HD-cT for preconditioning. It has been proposed that HD-cT results in a complete remission rate and progression-free survival (PFS) of 50%, compared with only 25% for CD-cT.22 In a retrospective analysis, HD-cT improved 2-year PFS versus CD-cT (50% vs. 28%, P <.001) and 5-year overall survival (53% vs. 41%, P <.001). Six adverse prognostic features can help identify candidates for HD-cT: extragonadal primary tumors; partial remission to first-line treatment; progression-free interval of 3 months or less; AFP elevation >1,000 ng/mL; beta-hCG elevation >1,000 U/L; and presence of liver, bone, or brain metastases.23 Findings from the ongoing phase III, international, randomized TIGER trial comparing HD-cT and CD-cT regimens should be considered in order to further guide treatment selection in this setting.24
If suboptimal response or relapse follows second-line chemotherapy, enrollment in a clinical trial is recommended. In patients not previously treated with CD-cT, HD-cT is an option.11 Relapse after HD-cT is generally considered incurable, and chemotherapy is administered with a palliative intent. In cisplatin-resistant tGCT or previous salvage treatment with HD-cT followed by stem-cell transplantation, gemcitabine and oxaliplatin are options based on GEMOX, a single-armed phase II study that demonstrated an overall response rate of 46% (95% CI, 30%-64%).25 Alternative third-line regimens with similar response rates include gemcitabine plus paclitaxel and single-agent high-dose oral etoposide. Although high-dose oral etoposide mitigates the need for port placement and prolonged clinic visits for infusions, the adverse effects can be significant, leading to pancytopenias and infections.26 Patients require routine laboratory work and frequent monitoring; therefore, careful patient selection is imperative.
Immunotherapy targeted against programmed death receptors has paved the way as a third-line treatment option for various solid tumors with microsatellite instability or mismatch repair testing (MSI-H/dMMR).11 The use of pembrolizumab in testicular cancer with MSI-H/dMMR is based on a single phase II clinical trial in 12 patients with nonseminoma tGCT that progressed after first-line cisplatin and second-line HD-cT or CD-cT. Pembrolizumab demonstrated little efficacy as monotherapy: Only two patients had stable disease, and no complete or partial responses were achieved at 28 weeks.27 Further trials are warranted to determine the role of immunotherapy in relapsed refractory tGCT.11
Adverse Effects
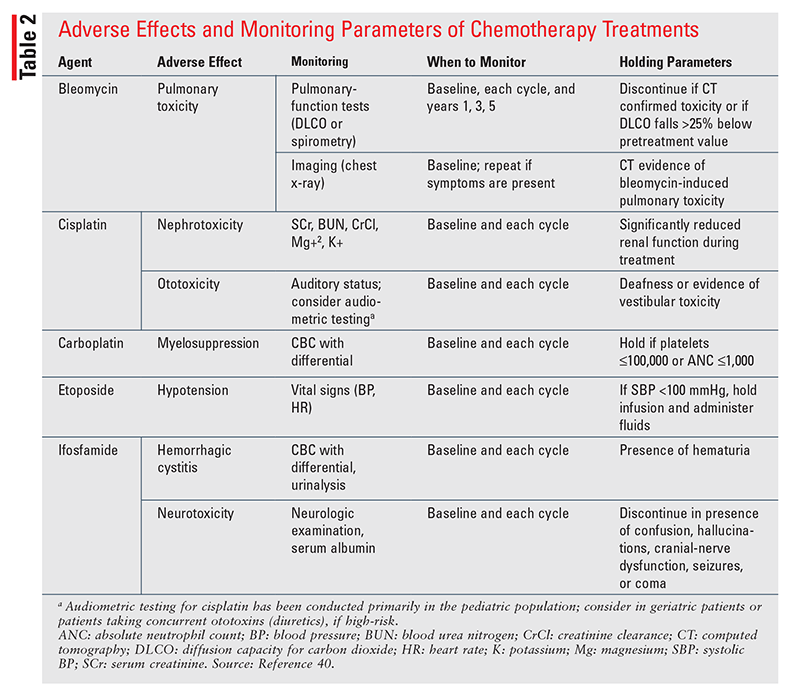
Cisplatin-based chemotherapy represents a break-through in the therapeutic landscape of tGCT, transforming it into a near-curable disease with a 10-year relapse-free survival rate of 89%.1 However, severe toxicities such as secondary malignancies, CV disease (CVD), neurotoxicity, nephrotoxicity, ototoxicity, pulmonary complications, and hypogonadism accompany this remarkable success. Treatment-related toxicities can emerge early or late after chemotherapy, surgery, or RT and can negatively impact overall quality of life in survivors.26 Therefore, it is imperative that pharmacists make every effort to mitigate and manage treatment-related adverse effects to minimize any potential impact on a patient’s physical or mental well-being. Baseline assessments and frequent monitoring are required for patients with tGCT who are undergoing active treatment (TABLE 2), and pharmacists play a pivotal role in monitoring for therapy-related adverse effects. The major adverse effects and the pharmacist’s role in mitigation strategies will be discussed in the following sections.
Acute Toxicities
Acute chemotherapy-related adverse effects include hypersensitivity reactions, nausea, vomiting, myelosuppression, and nephrotoxicity. Hypersensitivity can occur with any regimen, although those containing carboplatin and bleomycin carry the highest risk.26 Bleomycin-associated hypersensitivity reactions can manifest as life-threatening anaphylaxis with symptoms such as fever, bronchospasm, hypotension, and chills.28 To minimize the risk of hypersensitivity, a test dose of up to 2 U for the first two doses has been suggested to prime the immune system. If tolerated, the full dose may be administered for subsequent therapy. If a reaction is observed, abrupt discontinuation of the infusion is required, along with medical management of anaphylaxis, and rechallenge with bleomycin is not recommended. Carboplatin-induced hypersensitivity is unique in that it is not an immediate reaction, but rather occurs after repeated administration, most commonly after the 7th dose.26 Because the carboplatin regimen for tGCT is only two cycles, hypersensitivity is less likely to occur.
Nausea and vomiting are common adverse effects because all regimens use highly emetogenic chemotherapy agents, including cisplatin and carboplatin. All patients should be premedicated with antiemetics, with escalation of the regimen depending on patient tolerability.29 Myelosuppression is a predictable, dose-dependent toxicity from most agents, except for bleomycin.12 Owing to the highly curative nature of tGCT, delays in treatment due to myelosuppression are not recommended, and support with granulocyte colony-stimulating factor should be provided to minimize the risk of febrile neutropenia. Thrombocytopenia is a predominant hematologic toxicity with carboplatin, warranting close assessment for signs of bleeding.
Cisplatin accumulates within the renal tubules and causes cytotoxic damage, resulting in significant nephrotoxicity.30 Cisplatin-induced nephrotoxicity presents as an acute kidney injury in up to 20% to 30% of patients with preserved urine output and hypomagnesemia, although chronic kidney dysfunction may also occur.30 Volume expansion with normal saline is recommended to improve renal perfusion and prevent renal tubular accumulation. Aggressive hydration is administered to achieve a high urine output, with magnesium and potassium replacement added as needed. Amifostine, a cytoprotectant, has been studied for prevention of cisplatin-induced nephrotoxicity. However, current guidelines recommend its use only in patients with ovarian cancer or non–small-cell lung cancer receiving cisplatin dosages exceeding 100 mg/m2; it is not routinely recommended for patients with testicular cancer.31
Chronic Toxicities
Given that the average age for tGCT diagnosis is 33 years, patients may conceivably experience long-term morbidity of many years’ duration from treatment-related toxicities. Secondary neoplasms including leukemia, lymphoma, and solid tumors have been reported in testicular-cancer survivors, with an estimated 40-year cumulative incidence of about 30%.32 The risk of secondary leukemia from etoposide is dose dependent, with a 5-year incidence of 2.0% for dosages exceeding 2,000 mg/m2 versus 0.5% for dosages less than 2,000 mg/m2.33 RT alone carries a threefold higher risk of secondary malignancies than in patients not given RT.31 Secondary solid tumors can also occur and are most commonly located below the diaphragm, especially in patients given RT.32 Given the heightened risk of secondary malignancies in tGCT survivors, performance of routine site-specific cancer screening (colon, prostate, lung, if risk factors present) is paramount to ensure early detec- tion and management.33
CVD occurs in up to 16% of tGCT survivors and is one of the leading causes of long-term morbidity and mortality.4,6 CVD in tGCT survivors can manifest as coronary artery disease (CAD), myocardial infarction, hyperlipidemia, hypertension, obesity, and, in rare cases, Raynaud’s phenomenon.26 Low testosterone levels caused by treatment accelerate CVD and metabolic syndrome. The risk of developing CVD in tGCT treated with chemotherapy is sevenfold higher than in the general population.34 BEP treatment has been associated with a 5.7-fold increased risk of CAD (95% CI, 1.9-17.1) and a 3.1-fold increased risk of myocardial infarction (95% CI, 1.2-7.7).34 The mechanism of CVD remains poorly understood, but it has been proposed that direct vascular endothelial damage from cytokine release and oxidative damage during cytotoxic chemotherapy, along with mediastinal RT, plays an important role. No guidelines exist for CAD management in tGCT survivors specifically; however, clinicians should be aware of the increased CVD risk and employ early screening and risk-reducing strategies to minimize CVD-associated complications.34
Pneumonitis is a hallmark, dose-limiting toxicity of bleomycin that is life-threatening in up to 10% of patients.11 Bleomycin-induced pneumonitis (BIP) occurs secondary to a deficiency of bleomycin hydrolase, an enzyme responsible for bleomycin degradation and inactivation, in the lungs. Enzymatic deficiency of bleomycin hydrolase results in the accumulation of active bleomycin metabolites in the pulmonary tissue, along with free-radical production and cellular cytotoxicity. The time to onset of BIP is variable, ranging from a few weeks to years after treatment. Early symptoms are nonspecific and include exertional dyspnea and nonproductive cough. If BIP is left undiagnosed, there is a risk of progression to respiratory failure. Risk factors for BIP include cumulative doses exceeding 400 U, rapid IV infusion, supplemental oxygen, age exceeding 50 years, thoracic radiation, smoking, scuba diving, and concurrent use of granulocyte colony-stimulating factor.2,11 The most effective management strategy is to withhold bleomycin at the earliest signs or symptoms and mitigate the inflammatory response with corticosteroids, although minimal data support this approach.11 Bleomycin-free regimens should be considered in patients with underlying lung disease (i.e., chronic obstructive pulmonary disease), reduced glomerular filtration rate, and age exceeding 50 years.11 Monitoring parameters for bleomycin-induced pulmonary toxicity appear in TABLE 2.
Cisplatin-induced ototoxicity is a well-known dose- and duration-dependent phenomenon that can present months to years after treatment. Cisplatin causes selective damage to the outer hair cells of the cochlea that results in ototoxic symptoms, including tinnitus and hearing loss at high frequencies. The incidence of cisplatin-induced ototoxicity is estimated to be 10% to 40%, although this is likely underestimated owing to inherent challenges in the diagnosis.26 Vigilant monitoring of cumulative cisplatin doses is important for mitigating the risk of ototoxicity because 50% of patients receiving dosages greater than 400 mg/m2 will experience long-term persistent ototoxic sequelae.35 Other risk factors may include concurrent use of vinca alkaloids, preexisting hearing impairment, and rapid cisplatin infusion rates.
Cisplatin, along with ifosfamide and paclitaxel, confers a considerable risk of neurotoxicity in up to 76% of patients.27,36 Neurotoxicity can manifest as autonomic, sensory, motor, or peripheral neuropathy characterized by paresthesias, dysesthesias, motor disturbances, and vibratory sensations.27 Patients with preexisting diabetic neuropathy or other pain syndromes are more vulnerable to such toxicities. Neurotoxicity can arise after a single dose or after cumulative exposure. In severe cases, chemotherapy-induced neurotoxicity may lead to irreversible peripheral nerve damage and cognitive impairment, also termed “chemo brain.” The NCCN clinical practice guidelines recommend supportive care and duloxetine as preferred treatment when pharmacologic interventions are warranted.11 Ifosfamide causes a distinct neurotoxicity syndrome that closely resembles an acute, reversible encephalopathy syndrome that can occur in up to 10% of patients.26 Ifosfamide-induced neurotoxicity develops secondary to its metabolism into chloral hydrate, a neurotoxic metabolite that readily crosses the blood-brain barrier and causes oxidative stress. Because chloral hydrate is highly protein bound, hypoalbuminemia is a strong risk factor for ifosfamide-induced neurotoxicity. Other risk factors based on retrospective analysis include elevated serum creatinine and poor performance status.36 Treatment with methylene blue has been demonstrated to reverse the neurotoxic manifestations and may also be considered as a secondary prophylactic measure in patients requiring continued treatment.36
Fertility Preservation
Prior to determining a treatment course, a thorough discussion should take place with the patient regarding the potential for compromised fertility following tGCT treatment. This is especially prudent given that most patients are of reproductive age at the time of diagnosis. Various factors can impact fertility during and after cancer treatment. All primary treatment modalities increase the risk of infertility, but the degree varies based on the dose and intensity of RT or chemotherapy, the size and location of the RT field, surgery, and pretreatment hormonal insufficiency.37 Special attention should be paid to patients with pretreatment risks for infertility (e.g., atrophy of the contralateral testicle, family history of infertility).
The seminiferous tubules, where sperm are produced, are constantly undergoing active proliferation and differentiation. Cytotoxic chemotherapy used for testicular cancer readily targets this proliferative tissue; therefore, patients receiving treatment are most susceptible to fertility complications. Azoospermia can occur as rapidly as within the first 50 to 60 days after the first chemotherapy course.37 All patients should be strongly considered for fertility preservation, although the risk of prolonged or permanent azoospermia is drug-specific and dose-dependent. Platinum agents form the backbone of most chemotherapy regimens, and their broad cytotoxic effects pose a considerable risk of permanent azoospermia. A predictive factor for irreversible impairment of spermatogenesis is receipt of a cumulative dosage greater than 400 mg/m2.12 Cisplatin is dosed at 100 mg/m2 per cycle; therefore, most patients with stage I or early stage II disease who receive no more than one or two cycles are more likely to experience a return of fertility. In contrast, patients with advanced-stage disease who receive four or more cycles are deemed to be at highest risk. Compared with cisplatin, patients exposed to carboplatin have an improved probability of fertility after treatment.37 The alkylating agent used for testicular cancer, ifosfamide, poses the highest risk of permanent azoospermia regardless of total dose received.38
Guidelines set forth by the American Society of Clinical Oncology strongly recommend that patients receive information about fertility preservation and referral to a fertility specialist at the time of diagnosis.39 However, the lack of time for this discussion, perceived costs of fertility preservation, costs to the patient, and lack of convenient sperm-banking facilities are providers’ perceived barriers to routine counseling and triage to fertility care.37 The pharmacist can fulfill this unmet need by facilitating patient and provider education about the effect of chemotherapy on spermatogenesis and options for fertility preservation. Many online resources are available to direct patient discussion and referral, such as fertileHOPE, which provides information on cancer and infertility, fertility-preservation options, and locations offering fertility-preservation services, with additional personalized material for patient education.39 If a patient wishes to pursue fertility preservation, the primary modality offered is sperm cryopreservation (i.e., sperm banking). In this modality, viable sperm is collected before treatment is initiated in order to preserve it for future fertility plans.
The Pharmacist’s Role
Pharmacists can serve as an invaluable resource to facilitate the decision-making process to help improve quality of life after treatment of testicular cancer. By ensuring that patients receive guideline-directed therapy, appropriate monitoring of chemotherapy, and comprehensive education, pharmacists can assist patients in the decision regarding whether to pursue adjunctive chemotherapy and weigh it against the potential for acute and long-term treatment-related toxicities. For patients receiving chemotherapy, pharmacists play a valuable role in toxicity prevention and management. Pharmacists can screen medical and social histories to assess for specific risk factors that increase drug toxicity, such as bleomycin-induced pulmonary toxicity, cisplatin-induced nephrotoxicity, and ifosfamide-induced neurotoxicity. A comprehensive medication reconciliation and screening for concurrent medications that increase toxicity form an important toxicity-mitigation strategy to improve the tolerability of treatment (e.g., minimize nephrotoxin exposure with cisplatin or discontinue antihypertensives on etoposide). During initial and follow-up visits, pharmacists can inquire about the efficacy of the antiemetic regimen and modify it based on patient response. On the whole, pharmacists are essential members of the healthcare team who can provide comprehensive care to patients in order to maintain long-term survival rates with minimal toxicity.
REFERENCES
- American Cancer Society. Cancer facts & figures. www.cancer.org/ research/cancer-facts-statistics/all-cancer-facts-figures.html. Accessed March 16, 2020.
- Gilligan T, Lin DW, Aggarwal R, et al. Testicular cancer, version 2.2020, NCCN Clinical Practice Guidelines in Oncology. J Natl Compr Canc Netw. 2019;17:1529-1554.
- Daugaard G, Gundgaard MG, Mortensen MS, et al. Surveillance for stage I nonseminoma testicular cancer: outcomes and long-term follow-up in a population-based cohort. J Clin Oncol. 2014;32:3817-3823.
- Jones RH, Vasey PA. Part II: testicular cancer—management of advanced disease. Lancet Oncol. 2003;4:738-747.
- Williamson SR, Delahunt B, Magi-Galluzzi C, et al. The World Health Organization 2016 classification of testicular germ cell tumours: a review and update from the International Society of Urological Pathology Testis Consultation Panel. Histopathology. 2017;70:335-346.
- Cheng L, Albers P, Berney DM, et al. Testicular cancer. Nat Rev Dis Primers. 2018;4:29.
- Hemminki K, Chen B. Familial risks in testicular cancer as aetiological clues. Int J Androl. 2006;29:205-210.
- Ferguson L, Agoulnik AI. Testicular cancer and cryptorchidism. Front Endocrinol (Lausanne). 2013;4:32.
- Purdue MP, Devesa SS, Sigurdson AJ, McGlynn KA. International patterns and trends in testis cancer incidence. Int J Cancer. 2005;115:822-827.
- Daling JR, Doody DR, Sun X, et al. Association of marijuana use and the incidence of testicular germ cell tumors. Cancer. 2009;115:1215-1223.
- National Comprehensive Cancer Network. NCCN clinical practice guidelines in oncology (NCCN guidelines®). Testicular cancer. Version 2.2020. www.nccn.org/professionals/physician_gls/pdf/testicular.pdf. Accessed March 16, 2020.
- Amin MB, Edge S, Greene F, et al, eds. AJCC Cancer Staging Manual. 8th ed. New York, NY: Springer; 2017.
- International Germ Cell Consensus Classification: a prognostic factor-based staging system for metastatic germ cell cancers. International Germ Cell Cancer Collaborative Group. J Clin Oncol. 1997;15:594-603.
- Mazumdar M, Bajorin DF, Bacik J, et al. Predicting outcome to chemotherapy in patients with germ cell tumors: the value of the rate of decline of human chorionic gonadotrophin and alpha-fetoprotein during therapy. J Clin Oncol. 2001;19:2534-2541.
- Kollmannsberger C, Tandstad T, Bedard PL, et al. Patterns of relapse in patients with clinical stage I testicular cancer managed with active surveillance. J Clin Oncol. 2015;33:51-57.
- Dieckmann K-P, Dralle-Filiz I, Matthies C, et al. Testicular seminoma clinical stage 1: treatment outcome on a routine care level. J Cancer Res Clin Oncol. 2016;142:1599-1607.
- Yossepowitch O, Ehrlich Y, Lubin M, et al. Lymphovascular invasion in testicular germ cell tumors: clinicopathological correlates. Cent European J Urol. 2013;66:266-270.
- Tandstad T, Dahl O, Cohn-Cedermark G, et al. Risk-adapted treatment in clinical stage I nonseminomatous germ cell testicular cancer: the SWENO-TECA management program. J Clin Oncol. 2009;27:2122-2128.
- Chovanec M, Abu Zaid M, Hanna N, et al. Long-term toxicity of cisplatin in germ-cell tumor survivors. Ann Oncol. 2017;28:2670-2679.
- Culine S, Kerbrat P, Kramar A, et al. Refining the optimal chemotherapy regimen for good-risk metastatic nonseminomatous germ-cell tumors: a randomized trial of the Genito-Urinary Group of the French Federation of Cancers Centers (GETUG T93BP). Ann Oncol. 2007;18:917-924.
- van Dijk MR, Steyerberg EW, Habbema JDF. Survival of nonseminomatous germ cell cancer patients according to the IGCC classification: an update based on meta-analysis. Eur J Cancer. 2006;42:820-826.
- Feldman DR, Powles T. Salvage high-dose chemotherapy for germ cell tumors. Urol Oncol. 2015;33:355-362.
- Lorch A, Beyer J, Bascoul-Mollevi C, et al; International Prognostic Factors Study Group. Prognostic factors in patients with metastatic germ cell tumors who experienced treatment failure with cisplatin-based first-line chemotherapy. J Clin Oncol. 2010;28:4906-4911.
- Ongoing clinical trials in testicular cancer: the TIGER trial. Oncol Res Treat. 2016;39:553-556.
- Kollmannsberger C, Beyer J, Liersch R, et al. Combination chemotherapy with gemcitabine plus oxaliplatin in patients with intensively pretreated or refractory germ cell cancer: a study of the German Testicular Cancer Study Group. J Clin Oncol. 2004;22:108-114.
- Efstathiou E, Logothetis CJ. Review of late complications of treatment and late relapse in testicular cancer. J Natl Compr Canc Netw. 2006;4:1059-1070.
- Adra N, Einhorn LH, Althouse SK, et al. Phase II trial of pembrolizumab in patients with platinum refractory germ-cell tumors: a Hoosier Cancer Research Network Study GU14-206. Ann Oncol. 2018;29:209-214.
- Lam MSH. The need for routine bleomycin test dosing in the 21st century. Ann Pharmacother. 2005;39:1897-1902.
- National Comprehensive Cancer Network. NCCN clinical practice guidelines in oncology (NCCN guidelines®). Antiemesis. Version 1.2020. www. nccn.org/professionals/physician_gls/pdf/antiemesis.pdf. Accessed March 17, 2020.
- Fosså SD, Aass N, Winderen M, et al. Long-term renal function after treatment for malignant germ-cell tumours. Ann Oncol. 2002;13:222-228.
- Ethyol (amifostine) package insert. Nashville, TN: Cumberland Pharmaceuticals Inc; May 2017.
- Travis LB, Fosså SD, Schonfeld SJ, et al. Second cancers among 40,576 testicular cancer patients: focus on long-term survivors. J Natl Cancer Inst. 2005;97:1354-1365.
- Kollmannsberger C, Hartmann JT, Kanz L, Bokemeyer C. Therapy-related malignancies following treatment of germ cell cancer. Int J Cancer. 1999;83:860-863.
- Haugnes HS, Wethal T, Aass N, et al. Cardiovascular risk factors and morbidity in long-term survivors of testicular cancer: a 20-year follow-up study. J Clin Oncol. 2010;28:4649-4657.
- Lo Y, Shen L-J, Chen W-H, et al. Risk factors of ifosfamide-related encephalopathy in adult patients with cancer: a retrospective analysis. J Formos Med Assoc. 2016;115:744-751.
- Mulhall JP, Applegarth LD, Oates RD, Schlegel PN. Fertility Preservation in Male Cancer Patients. New York, NY: Cambridge University Press; 2013.
- Ping P, Gu B-H, Li P, et al. Fertility outcome of patients with testicular tumor: before and after treatment. Asian J Androl. 2014;16:107-111.
- Oktay K, Harvey BE, Loren AW. Fertility preservation in patients with cancer: ASCO Clinical Practice Guideline Update Summary. J Oncol Pract. 2018;14:381-385.
- NCCN chemotherapy order templates (NCCN templates®). www.nccn. org/professionals/OrderTemplates/Default.aspx. Accessed March 17, 2020.