The Treatment of Depression in Parkinson's Disease
RELEASE DATE
May 1, 2023
EXPIRATION DATE
May 31, 2025
FACULTY
Brooke Barlow, PharmD
Neurocritical Care Clinical Pharmacy Specialist
Memorial Hermann The Woodlands Medical Center
The Woodlands, Texas
FACULTY DISCLOSURE STATEMENTS
Dr. Barlow has no actual or potential conflicts of interest in relation to this activity.
Postgraduate Healthcare Education, LLC does not view the existence of relationships as an implication of bias or that the value of the material is decreased. The content of the activity was planned to be balanced, objective, and scientifically rigorous. Occasionally, authors may express opinions that represent their own viewpoint. Conclusions drawn by participants should be derived from objective analysis of scientific data.
ACCREDITATION STATEMENT
 Pharmacy
Pharmacy
Postgraduate Healthcare Education, LLC is accredited by the Accreditation Council for Pharmacy Education as a provider of continuing pharmacy education.
UAN: 0430-0000-23-054-H01-P
Credits: 2.0 hours (0.20 ceu)
Type of Activity: Knowledge
TARGET AUDIENCE
This accredited activity is targeted to pharmacists. Estimated time to complete this activity is 120 minutes.
Exam processing and other inquiries to:
CE Customer Service: (800) 825-4696 or cecustomerservice@powerpak.com
DISCLAIMER
Participants have an implied responsibility to use the newly acquired information to enhance patient outcomes and their own professional development. The information presented in this activity is not meant to serve as a guideline for patient management. Any procedures, medications, or other courses of diagnosis or treatment discussed or suggested in this activity should not be used by clinicians without evaluation of their patients' conditions and possible contraindications or dangers in use, review of any applicable manufacturer's product information, and comparison with recommendations of other authorities.
GOAL
To review the pathophysiology, presentation, diagnosis, and treatment of depression in Parkinson's disease (PD) and explain the pharmacist's role in developing a patient-centered pharmacotherapy plan.
OBJECTIVES
After completing this activity, the participant should be able to:
- Explain the pathophysiology of depression in PD.
- Describe the clinical presentation of and diagnostic criteria for depression in PD.
- Identify nonpharmacologic and pharmacologic treatments for depression in PD.
- Discuss the pharmacist's role in the pharmacotherapeutic care of PD patients with depression.
ABSTRACT: Although it is frequently characterized as a movement disorder, it has been suggested that Parkinson’s disease (PD) may be more accurately considered a neuropsychiatric disorder. Depression is the most common mental health disorder in patients with PD; however, effective treatment remains poorly defined. Untreated or inadequately treated depression in PD can result in motor-symptom exacerbation, increased disability, impaired quality of life, and worse outcomes. Nonpharmacologic treatment improves symptoms, but when used alone it may be insufficient for severe depression. A variety of antidepressants are used to treat depression in PD. Because medication regimens for PD can be complex, the pharmacist’s knowledge of potential drug-drug interactions, adverse effects, and contraindications associated with antidepressants is critically important for reducing the risk of adverse effects.
Parkinson’s disease (PD) is often characterized as a primary movement disorder; however, nonmotor symptoms—notably, neuropsychiatric complications—have gained recognition as common and disabling features of PD. The high prevalence of neuropsychiatric manifestations in PD suggests that the disease may be more accurately characterized as a neuropsychiatric disorder.1 Neuropsychiatric manifestations can include anxiety, depression, apathy, psychosis, and sleep disturbances.2 Depression is the most common mental health disorder in PD, occurring up to 50% of patients.3 Proposed risk factors for depression in PD include female sex, family history of depression, early-onset PD, atypical parkinsonism, and presence of other psychiatric comorbidities.1 The severity of depression in PD varies: An estimated 5% to 20% of patients have major (severe) depression, and 10% to 30% have minor forms.4 Severe motor fluctuations can further exacerbate depressive symptoms.5
Despite the high prevalence, depression in PD is undiagnosed and untreated in up to 60% of patients, leading to reduced quality of life. Untreated depression can result in an inability to perform activities of daily living, increased need for symptomatic PD therapy, and decreased medication adherence, which further exacerbates the disease.6 Pharmacists play a critical role in assessing PD patients for depression and recommending a tailored pharmacotherapeutic plan to control symptoms while reducing unwanted side effects or potential drug interactions from therapy. This article will review the pathophysiology, clinical presentation, diagnosis, and nonpharmacologic and pharmacologic treatments for depression in PD and discuss the pharmacist’s role in developing a patient-centered pharmacotherapy plan.
Pathophysiology
The pathophysiologic hallmark of PD is the loss of dopaminergic neurons in the substantia nigra and intraneuronal Lewy bodies, which leads to the cardinal features of bradykinesia, resting tremor, rigidity, and postural instability.7 Loss of dopaminergic neurotransmission is thought to extend beyond the midbrain and disrupt serotonergic and noradrenergic neurotransmission in the orbitofrontal–basal ganglia–thalamic circuits.8 Because these neuronal systems are critical to mood regulation, aberrations of these pathways, as seen in PD, can induce depression and other psychiatric disturbances.
Beyond the neurodegenerative hypothesis for PD-related depression, PD treatments may also contribute to the pathogenesis of depression. Deep brain stimulation (DBS) is considered a last-line treatment in patients with advanced PD to control refractory motor symptoms despite maximized medical therapy. Following implantation of the DBS device, mood disturbances (including depression) have been observed after subthalamic or substantia nigra stimulation.9 Abrupt withdrawal from dopamine agonist therapy (e.g., pramipexole, ropinirole, rotigotine) can also result in profound disability, with exacerbations of motor symptoms as well as nonmotor symptoms including depression and anxiety.10 Therefore it is critically important for pharmacists to counsel patients not to abruptly discontinue their medications and to ensure prompt resumption of these therapies in the inpatient setting.
Clinical Presentation and Diagnosis
In patients with PD, depression can be difficult to diagnose because its features overlap with those of PD itself. The fatigue, blunted affect, and cognitive slowing occurring in depression can be difficult to differentiate from bradykinesia and masked facies (i.e., reduced facial expressivity), which are classic features of PD. Apathy (characterized by diminished motivation), which is commonly seen in PD, can also resemble depressive symptoms; the primary distinguishing factor is mood, which is negative in depression versus neutral in apathy.11 No specific screening or diagnostic tool has been developed to distinguish between these overlapping symptoms in patients with PD.
A systematic review was conducted to clarify evidence-based guidelines for the diagnosis and treatment of depression in PD.12 According to the review, a National Institute of Neurological Disorders and Stroke committee stated that to avoid the potential misdiagnosis of depression as apathy, depressed mood (rather than just decreased interest) must be present, and recommended that screening for depression in PD be performed during the “on” phase (i.e., PD symptoms well-controlled). Furthermore, to avoid false-negatives, it was recommended that all symptoms be assessed for depression regardless of whether they may be related to the motor disorder.12
The self-rated Patient Health Questionnaire-9 (PHQ-9) and Geriatric Depression Scale are recommended for initial screening of depression because of their efficiency, extensive use in research, and brevity of symptom assessment.12 In a cohort of patients with PD, the PHQ-9 had 100% sensitivity and 83% specificity for detecting major depression.12 The criteria of the American Psychiatric Association’s Diagnostic and Statistical Manual of Mental Disorders, Fifth Edition, are the standard for diagnosing major depressive disorder (TABLE 1); however, they lack specificity and validation for PD.12 Additional features of the initial assessment of PD patients who present with depressive symptoms include a comprehensive medical/psychiatric history, medication review, and laboratory screening (e.g., CBC) to rule out alternative diagnoses, such as anemia, hypothyroidism, and vitamin B12 deficiency. Depending on symptom severity or clinical suspicion of other etiologies, neuroimaging, toxicology screening, HIV testing, or neurology consultation may be considered.

Nonpharmacologic Treatment
The treatment of depression in PD requires a comprehensive multidimensional approach, including psychotherapy (to learn skills to effectively cope with depressive symptoms and mitigate stressors) and education (on expectations concerning time to, and degree of, symptom resolution). Exercise, healthy diet, adequate sleep hygiene, and social support are lifestyle modifications that can be effective for alleviating depressive symptoms. Other nonpharmacologic interventions include cognitive-behavioral therapy (CBT), electroconvulsive therapy (ECT), meditation, and repetitive transcranial magnetic stimulation (rTMS).13
CBT appears to be safe and efficacious for use in PD, although it may be insufficient when the depression is more severe.14 In trials, ECT used for depression in PD demonstrated a 93.1% improvement in depressive symptoms and an 83% improvement in motor symptoms; however, ECT is also associated with delirium and confusion, necessitating discontinuation in some patients.15 In rTMS, a high-frequency stimulus is delivered to cortical regions of the brain, inducing an excitatory or inhibitory response.16 Evidence on rTMS’s benefits has been mixed, with some studies demonstrating efficacy similar to that of antidepressants and reporting improvements in motor symptoms; however, other studies have not found a benefit compared with placebo.12
Pharmacologic Treatment
Pharmacotherapy for depression in PD comprises antidepressants including selective serotonin reuptake inhibitors (SSRIs), serotonin-norepinephrine reuptake inhibitors (SNRIs), tricyclic antidepressants (TCAs), monoamine oxidase type B (MAO-B) inhibitors, and bupropion (TABLE 2). However, prior to antidepressant initiation, it is essential to ensure that the patient’s motor symptoms are adequately controlled because of the correlation between the “off” phase (i.e., PD-symptom recurrence) and depressive symptoms.17 If treatment of PD symptoms is necessary, it would be advantageous to initiate an MAO-B inhibitor to address both motor and depressive symptoms. If it is decided to pursue antidepressant therapy, the optimal agent remains poorly defined. SSRIs and TCAs have been most studied to date, with a smaller number of trials conducted on SNRIs.18 A meta-analysis of antidepressant therapy in PD found that all agents were beneficial compared with placebo, but when the agents were stratified by class, only SSRIs demonstrated a significant effect.16 In contrast, other studies have found TCAs to confer the greatest benefit. Treatment should be based on an assessment of the potential benefits and adverse effects (AEs) or drug interactions for the individual patient.
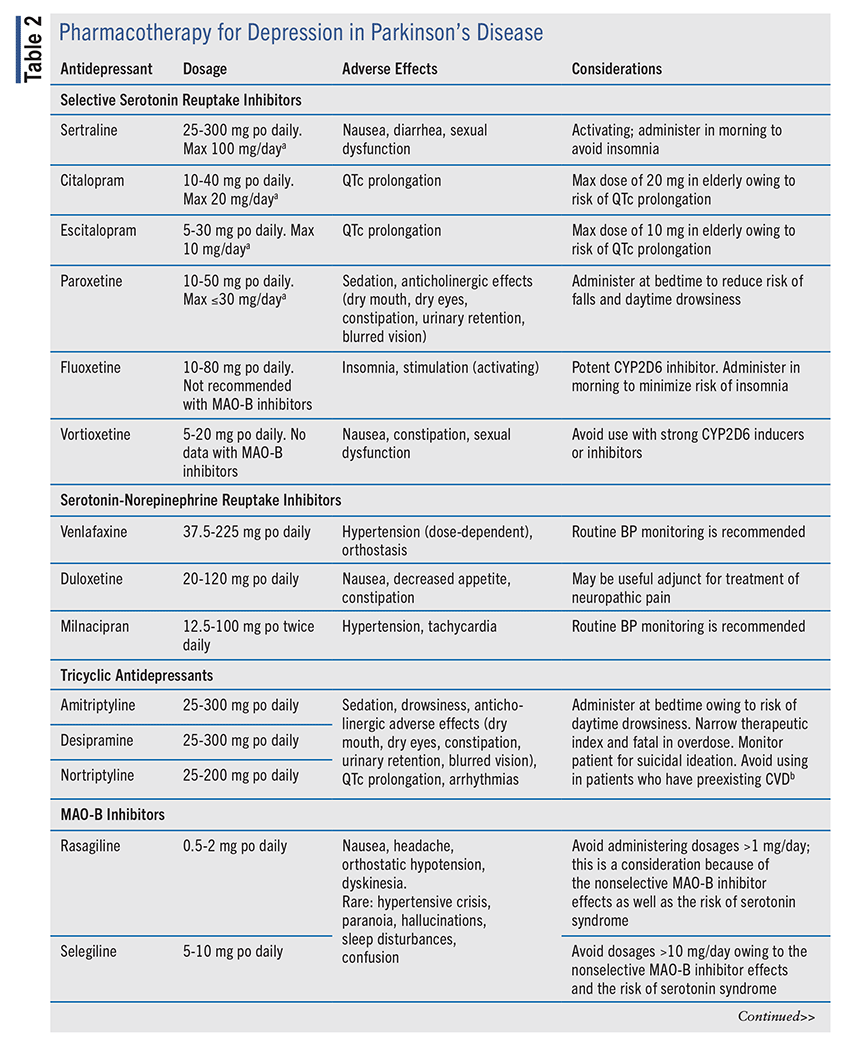

SSRIs: SSRIs exert their antidepressant effects by inhibiting presynaptic serotonin reuptake, which increases the availability of serotonin in the synaptic cleft to potentiate neurotransmission. Currently, seven SSRIs are FDA approved for depression: citalopram, escitalopram, fluoxetine, paroxetine, sertraline, vilazodone, and vortioxetine. Vortioxetine and vilazodone have additional unique mechanisms as agonists of 5HT1A and/or 5HT1B receptors.19 SSRIs are often employed as first-line treatment for depression because of their tolerability and limited drug-drug interactions.
Agents evaluated in randomized, controlled trials of SSRIs for PD include citalopram, escitalopram, fluoxetine, paroxetine, and sertraline.20-24 Although early studies showed no effect, a review of recent trials of SSRIs demonstrated a significant reduction in standardized depression scores compared with placebo.25 Follow-up duration in these studies was around 8 to 12 weeks, with therapy benefit occurring within 4 weeks of continued therapy. No study demonstrated a complete resolution of symptoms.25 Vortioxetine, one of the newer FDA-approved agents, was evaluated in a prospective, open-label, single-arm trial including 30 patients with PD and demonstrated a 50% reduction in the 17-item Hamilton Depression Rating Scale (HAM-D17) at 12 weeks.26 Additionally, symptoms of apathy, cognition, fatigue, and quality of life were significantly improved at 3 weeks.26 SSRIs are generally well tolerated, although gastrointestinal AEs were the most common reason for treatment discontinuation.25,27 Some studies reported worsening bradykinesia and extrapyramidal symptoms following citalopram initiation, but other studies did not.22,24,27,28
Selection of an antidepressant for PD should consider the agents’ relative AE profiles. Paroxetine has a high affinity for muscarinic receptors, which may exacerbate dry mouth, constipation, urinary retention, cognitive impairment, and drowsiness; it also has a short half-life and can rapidly induce withdrawal syndrome upon sudden discontinuation.29 In contrast, fluoxetine can be activating and induce agitation; its long half-life also leads to a slower initial response of antidepressant effects due to the time to achieve steady-state concentrations, but the prolonged duration minimizes the risk of withdrawal upon discontinuation.29 Citalopram and escitalopram can prolong the QTc interval, so caution and ECG monitoring are advised when initiating these agents in patients with a history or current use of other QTcprolonging medications.30 In patients with prolonged QTc requiring SSRI therapy, fluoxetine, fluvoxamine, and sertraline appear to have a low risk for QTc prolongation, and paroxetine carries the lowest risk.31 Sertraline appears to be well tolerated overall.
Evaluation for potential drug-drug interactions with SSRI initiation is critical to lower the risk of toxicities. Fluoxetine, a potent CYP2D6 inhibitor, results in increased concentrations of 2D6 substrates (e.g., bupropion, metoprolol, risperidone). Fluvoxamine is a strong inhibitor of CYP1A2 and CYP2C19 and a moderate inhibitor of CYP2C9 and CYP3A4, and it can impact several high-risk, narrow-therapeutic-index medications, including warfarin, theophylline, methadone, and clozapine.32 All SSRIs can potentiate the risk of serotonin syndrome if they are administered concurrently with other serotonergic agents (e.g., SNRIs, TCAs, trazodone) or MAO type A (MAO-A) or nonselective MAO inhibitors (e.g., linezolid, tranylcypromine, phenelzine, methylene blue).
SNRIs: Venlafaxine, desvenlafaxine, duloxetine, milnacipran, and levomilnacipran are FDA approved for depression. SNRIs are proposed to reduce depressive symptoms by inhibiting serotonin and norepinephrine reuptake into the presynaptic neuron, thereby increasing the availability of these neurotransmitters within the synaptic cleft; milnacipran and levomilnacipran preferentially inhibit norepinephrine reuptake. SNRIs can be advantageous for depression in PD because of their dual antidepressant and analgesic effects.
A randomized, controlled trial of extended-release venlafaxine found a significant reduction in HAM-D score at 12 weeks, with no negative impact on motor symptoms.24 In an open-label, multicenter, single-arm trial, duloxetine 60 mg daily significantly reduced HAMD-17 and other depression scoring measures during the treatment period, with no observed worsening of PD-related tremor.33 Despite duloxetine’s efficacy for neuropathic pain in other populations, a randomized, controlled trial of duloxetine 40 mg for 10 weeks in PD patients failed to show a significant reduction in pain severity as assessed by the visual analogue scale; however, it remains to be determined whether higher doses are needed for effective pain relief in PD.34 Data assessing milnacipran for use in depression in PD are limited, with a single case report demonstrating depressive-symptom relief in patients who had an insufficient response to SSRIs.35
SNRIs are well tolerated overall, and the most common AEs are gastrointestinal (e.g., nausea, diarrhea). However, owing to their effects on the sympathetic nervous system, SNRIs’ AE profile differs from that of SSRIs. Norepinephrine potentiation results in a dose-dependent increase in blood pressure (BP), with sustained elevations of 15 mmHg occurring in up to 12% of PD patients taking venlafaxine.24 At low doses (<200 mg/ day), venlafaxine preferentially inhibits serotonin reuptake, whereas doses >225 mg result in preferential inhibition of norepinephrine reuptake.36 The extended-release form is thought to have a lower risk of hypertension than the immediate-release form; however, orthostatic hypotension is threefold higher with its use.37 This is of critical importance in PD patients who frequently experience orthostasis. Therefore, SNRIs should be used with caution in PD patients with comorbid hypertension, cardiovascular (CV) disease, or significant orthostasis.
TCAs: In addition to their advantages in treating neuropathic pain and insomnia and preventing tension-type headaches, TCAs remain a mainstay therapy for depression. The proposed antidepressive effects of TCAs are secondary to the inhibition of norepinephrine and serotonin reuptake in the presynaptic cleft, but TCAs exhibit several off-target effects through blockade of histamine, adrenergic, and muscarinic-acetylcholine receptors.38 TCAs possess a three-ringed structure that is attached to a secondary or tertiary amine. Secondary amines include desipramine and nortriptyline, which are thought to have greater norepinephrine blockade in comparison with the greater serotonergic effect of tertiary amines (e.g., amitriptyline, imipramine, doxepin). In one study, nortriptyline was more effective than paroxetine or placebo, and another study had similar results for desipramine versus citalopram and placebo; however, AEs including dry mouth, constipation, hyperhidrosis, hot flashes, and dysuria were twofold higher with desipramine.21,27 A meta-analysis suggested that TCAs may be more effective than SSRIs for depression in PD; however, evidence is conflicting.12
TCAs are less tolerated than SSRIs and SNRIs because of their off-targeted AEs on muscarinic and histaminergic receptors and sodium channel blockade. Anticholinergic effects are most prominent with the tertiary amines such as amitriptyline and appear to be less pronounced with the secondary amines such as nortriptyline and desipramine.39 CV toxicity with TCAs can be severe and life-threatening, especially in overdose situations. TCAs are arrhythmogenic secondary to potent blockade of sodium and potassium channels. Other CV effects of TCAs include QTc-interval prolongation, orthostatic hypotension, and suppression of myocardial contractility, and TCAs have been linked to cases of worsening ischemic heart disease and sudden cardiac death.40 TCAs should be avoided in patients who have significant CV disease or are at risk for severe arrythmias as well as in patients with narrow angle-closure glaucoma, owing to the risk of increased intraocular pressure.12 The sedative effects of TCAs are more pronounced than those for other antidepressants and heighten the risk for falls and delirium; therefore caution should be exercised in PD patients with concurrent dementia or cognitive impairment.12 Because of their high lethality index, TCAs should be avoided in patients with suicidal ideation or a history of suicide attempts.
MAO-B Inhibitors: Frequently used for symptomatic relief of motor symptoms in PD, MAO-B inhibitors are also beneficial for comorbid depression. The three FDA-approved MAO-B inhibitors—rasagiline, selegiline, and safinamide—work centrally by inhibiting the MAO-B enzyme, which is responsible for dopamine oxidation (i.e., breakdown). Inhibition of this enzyme increases dopamine concentrations in the synaptic gap. Selegiline and rasagiline exhibit dose-dependent effects on MAO inhibition; low doses used for PD exhibit selective MAO-B inhibition, and high doses (>10 mg/day, >1 mg/day) exert nonselective MAO-B and MAO-A inhibition.41 In a meta-analysis of six randomized, controlled trials, MAO-B inhibitors significantly reduced the severity of depressive symptoms.42 Subgroup analysis revealed the greatest benefit for antidepressive effects in early-stage PD and with short-term treatment (90-120 days), but not with long-term treatment (>24 weeks) or in middle- or late-stage PD.42 More trials are needed to determine the efficacy of MAO-B inhibitors in more advanced PD.
As mentioned previously, selegiline and rasagiline exhibit nonselective MAO inhibition at higher doses than are used for PD. MAO-A inhibition decreases the breakdown of serotonin and norepinephrine, which can be problematic with concurrent serotonergic medications owing to the potentiation of serotonin syndrome.43 Although package labeling states that SSRIs, SNRIs, and TCAs are contraindicated with MAO-B inhibitor use, postmarketing studies suggest that this risk is likely overstated and dependent on the type and number of agents used.44
A retrospective, multicenter, cohort study of 1,504 patients taking concurrent SSRIs (10% taking >1 antidepressant) found no cases of serotonin syndrome.45 A subgroup analysis of the ADAGIO study, which evaluated rasagiline versus placebo in de novo PD, found a significant improvement in nonmotor symptoms and no cases of serotonin syndrome in the 16.3% of patients taking concurrent antidepressants.46 In a survey analysis of 4,568 PD patients, the incidence of possible serotonin syndrome was 0.24% (0.04% considered serious) with selegiline and concurrent antidepressant use.47 Nearly all of the cases of serotonin syndrome with selegiline occurred with concurrent fluoxetine use at dosages ≥20 mg/day.47
There are strict dosing restrictions with concurrent serotonergic agents (e.g., sertraline ≤100 mg, paroxetine ≤30 mg, escitalopram ≤10 mg, citalopram ≤20 mg).46 The following points are critically important: 1) Do not exceed the maximum recommended dosage of MAO-B inhibitors for PD if the patient is taking concurrent serotonergic medications; 2) SSRIs should be initiated at the lowest dose possible and slowly titrated; 3) do not exceed maximum recommended SSRI doses; 4) avoid concurrent use of medications that have drug interactions with SSRIs or MAO-B inhibitors; and 5) routinely screen medication profiles and assess patients for signs and symptoms of serotonin when MAO-B inhibitors and serotonergic medications are used concurrently.44
Other Antidepressants: Bupropion is different from other antidepressants in that it does not target serotonin; instead, it inhibits the reuptake of dopamine and norepinephrine. Theoretically, the dopaminergic effects may improve both motor and neuropsychiatric effects of PD; however, the evidence is conflicting. Data on bupropion for depression in PD are limited to case reports and observational studies, with a single randomized trial demonstrating considerable efficacy for depression and compulsive behavior.48 Loss of dopaminergic neurotransmission is proposed to cause apathy, for which bupropion has shown positive effects.49 Although evidence is conflicting, some cases of worsening psychosis have been reported in PD patients taking bupropion; therefore its use is cautioned in PD with concurrent psychosis/psychiatric condition.50
Mirtazapine has multiple mechanisms, including central blockade of noradrenergic reuptake, blockade of 5HT2A and 5HT2C, and blockade of histamine receptors (H1). Limited data are available regarding the use of mirtazapine in depression; however, this agent has been shown to be useful for improving visual hallucinations and psychosis in PD.51 Mirtazapine may also be helpful for improving sleep quality and appetite given the H1blockade. The use of trazodone, another atypical antidepressant, is not recommended because of the risk of worsening parkinsonism.52,53
The Pharmacist's Role
Pharmacists are essential members of the multidisciplinary team in developing appropriate, safe, and effective pharmacotherapy plans for PD patients with depression. In the initial medication assessment, the pharmacist should inquire about adherence to PD medications, as motor symptoms should be controlled prior to antidepressant initiation. Encouraging patients to keep a journal of depressive symptoms, potential exacerbating features, and presence or absence of PD motor symptoms can be helpful in guiding adjustments of PD medications or antidepressant therapy.17 In general, initiation of an SSRI is a reasonable first-line choice given the improved tolerability compared with other antidepressants.20 However, the decision should be personalized based on the presence of concurrent conditions wherein a different antidepressant may be more suitable (e.g., duloxetine for neuropathic pain).
Selection of the optimal antidepressant must consider the risk of AEs that can exacerbate PD symptoms. The anticholinergic and CV effects of TCAs may be intolerable in patients with advanced PD given the high incidence of dementia and orthostasis, respectively. For PD patients who are prescribed medications that can be sedating (e.g., TCAs, paroxetine), pharmacists can recommend bedtime administration to minimize daytime drowsiness and the risk of falls. Conversely, for patients prescribed medications with a stimulatory effect (e.g., fluoxetine, MAO-B inhibitors), morning administration may be recommended to minimize insomnia. Medication adherence should also be taken into account; e.g., in patients with predicted poor adherence, agents with a longer duration of action (i.e., fluoxetine) should be considered over shorter-acting agents (e.g., paroxetine, venlafaxine) to minimize serotonin withdrawal syndrome.
Appropriate monitoring of the selected antidepressant is vital to ensure safe use during titration and maintenance. Examples include BP monitoring during venlafaxine therapy; ECG monitoring with TCAs, escitalopram, or citalopram; and assessment for signs and symptoms of serotonin syndrome with MAO-B inhibitors or SSRIs. It is crucial for the pharmacist to complete a comprehensive medication review to lessen the risk of drug-drug interactions with antidepressant therapy. Pharmacists should screen medication profiles for other serotonergic-acting medications or MAO inhibitors and make recommendations for alternative therapies to minimize the risk of serotonin syndrome. For patients taking medications metabolized via CYP450 pathways, it is essential to recommend avoidance of agents that are potent inhibitors of these enzymes (e.g., fluoxetine, fluvoxamine) and suggest alternatives.
Patients taking MAO-B inhibitors should be advised to limit their intake of tyramine-rich foods (e.g., cheese, red wine, chocolate).54 Tyramine is a monoamine precursor and can generate surges of catecholamines upon MAO inhibition, resulting in a rapid increase in heart rate and BP.54 Bupropion should be avoided in patients with epilepsy or a history of eating disorders. Patients and caregivers should be educated about dietary modifications and the signs and symptoms of serotonin syndrome, especially if the patient is taking concurrent MAO-B inhibitors and antidepressants.
Conclusion
Depression is a common comorbidity in patients with PD, but it often goes undiagnosed because of the symptom overlap between the two conditions. Pharmacists should perform routine depression screening in patients with PD and, when necessary, facilitate the selection of medications. Nonpharmacologic management of depression, including healthy-lifestyle modifications, improved sleep, and daily activity, is important for symptom improvement. Antidepressant selection in patients with PD requires a personalized approach based on differences in the various agents’ mechanisms of action, AEs, and drug- or food-interaction profiles. Pharmacists play a vital role in optimizing medication selection for depression in PD and ensuring that the underlying motor symptoms are controlled so that depression exacerbations are kept to a minimum.
REFERENCES
1. Weintraub D, Burn DJ. Parkinson’s disease: the quintessential neuropsychiatric disorder. Mov Disord. 2011;26(6):1022-1031.
2. Weintraub D, Mamikonyan E. The neuropsychiatry of Parkinson disease: a perfect storm. Am J Geriatr Psychiatry. 2019;27(9):998-1018.
3. Reijnders JS, Ehrt U, Weber WE, et al. A systematic review of prevalence studies of depression in Parkinson’s disease. Mov Disord. 2008;23(2):183-189; quiz 313.
4. Allain H, Schuck S, Mauduit N. Depression in Parkinson’s disease. BMJ. 2000;320(7245):1287-1288.
5. Ossig C, Sippel D, Fauser M, et al. Timing and kinetics of nonmotor fluctuations in advanced Parkinson’s disease. J Parkinsons Dis. 2017;7(2):325-330.
6. Ravina B, Camicioli R, Como PG, et al. The impact of depressive symptoms in early Parkinson disease. Neurology. 2007;69(4):342-347.
7. Marsh L. Depression and Parkinson’s disease: current knowledge. Curr Neurol Neurosci Rep. 2013;13(12):409.
8. Mayberg HS, Solomon DH. Depression in Parkinson’s disease: a biochemical and organic viewpoint. Adv Neurol. 1995;65:49-60.
9. Zarzycki MZ, Domitrz I. Stimulation-induced side effects after deep brain stimulation—a systematic review. Acta Neuropsychiatr. 2020;32(2):57-64.
10. Rabinak CA, Nirenberg MJ. Dopamine agonist withdrawal syndrome in Parkinson disease. Arch Neurol. 2010;67(1):58-63.
11. Richard IH. Apathy does not equal depression in Parkinson disease: why we should care. Neurology. 2006;67(1):10-11.
12. Starkstein SE, Brockman S. Management of depression in Parkinson’s disease: a systematic review. Mov Disord Clin Pract. 2017;4(4):470-477.
13. Pontone GM, Mills KA. Optimal treatment of depression and anxiety in Parkinson’s disease. Am J Geriatr Psychiatry. 2021;29(6):530-540.
14. Troeung L, Egan SJ, Gasson N. A meta-analysis of randomised placebo-controlled treatment trials for depression and anxiety in Parkinson’s disease. PloS One. 2013;8(11):e79510.
15. Borisovskaya A, Bryson WC, Buchholz J, et al. Electroconvulsive therapy for depression in Parkinson’s disease: systematic review of evidence and recommendations. Neurodegener Dis Manag. 2016;6(2):161-176.
16. Bomasang-Layno E, Fadlon I, Murray AN, Himelhoch S. Antidepressive treatments for Parkinson’s disease: a systematic review and meta-analysis. Parkinsonism Relat Disord. 2015;21(8):833-842; discussion 833.
17. Timmer MH, van Beek MH, Bloem BR, Esselink RA. What a neurologist should know about depression in Parkinson’s disease. Pract Neurol. 2017;17(5):359-368.
18. Ryan M, Eatmon CV, Slevin JT. Drug treatment strategies for depression in Parkinson disease. Expert Opin Pharmacother. 2019;20(11):1351-1363.
19. Wagner G, Schultes MT, Titscher V, et al. Efficacy and safety of levomilnacipran, vilazodone and vortioxetine compared with other second-generation antidepressants for major depressive disorder in adults: a systematic review and network meta-analysis. J Affect Disord. 2018;228:1-12
20. Leentjens AF, Vreeling FW, Luijckx GJ, Verhey FR. SSRIs in the treatment of depression in Parkinson’s disease. Int J Geriatr Psychiatry. 2003;18(6):552-554.
21. Menza M, Dobkin RD, Marin H, et al. A controlled trial of antidepressants in patients with Parkinson disease and depression. Neurology. 2009;72(10):886-892.
22. Wermuth L, Sørensen PS, Timm S, et al. Depression in idiopathic Parkinson’s disease treated with citalopram: a placebo-controlled trial. Nord J Psychiatry. 1998;52(2):163-169.
23. Weintraub D, Taraborelli D, Morales KH, et al. Escitalopram for major depression in Parkinson’s disease: an open-label, flexible-dosage study. J Neuropsychiatry Clin Neurosci. 2006;18(3):377-383.
24. Richard IH, McDermott MP, Kurlan R, et al. A randomized, doubleblind, placebo-controlled trial of antidepressants in Parkinson disease. Neurology. 2012;78(16):1229-1236.
25. Prange S, Klinger H, Laurencin C, et al. Depression in patients with Parkinson’s disease: current understanding of its neurobiology and implications for treatment. Drugs Aging. 2022;39(6):417-439.
26. Santos García D, Alonso Losada MG, Cimas Hernando I, et al. Vortioxetine improves depressive symptoms and cognition in Parkinson’s disease patients with major depression: an open-label prospective study. Brain Sci. 2022;12(11):1466.
27. Devos D, Dujardin K, Poirot I, et al. Comparison of desipramine and citalopram treatments for depression in Parkinson’s disease: a double-blind, randomized, placebo-controlled study. Mov Disord. 2008;23(6):850-857.
28. Montastruc JL, Fabre N, Blin O, et al. Does fluoxetine aggravate Parkinson’s disease? A pilot prospective study. Mov Disord. 1995;10(3):355-357.
29. Anderson IM, Edwards JG. Guidelines for choice of selective serotonin reuptake inhibitor in depressive illness. Adv Psychiatr Treat. 2001;7(3):170-180.
30. Funk KA, Bostwick JR. A comparison of the risk of QT prolongation among SSRIs. Ann Pharmacother. 2013;47(10):1330-1341.
31. Beach SR, Kostis WJ, Celano CM, et al. Meta-analysis of selective serotonin reuptake inhibitor-associated QTc prolongation. J Clin Psychiatry. 2014;75(5):e441-e449.
32. Spina E, Scordo MG. Clinically significant drug interactions with antidepressants in the elderly. Drugs Aging. 2002;19(4):299-320.
33. Bonuccelli U, Meco G, Fabbrini G, et al. A non-comparative assessment of tolerability and efficacy of duloxetine in the treatment of depressed patients with Parkinson’s disease. Expert Opin Pharmacother. 2012;13(16):2269-2280.
34. Iwaki H, Ando R, Tada S, et al. A double-blind, randomized controlled trial of duloxetine for pain in Parkinson’s disease. J Neurol Sci. 2020;414:116833.
35. Takahashi H, Kamata M, Yoshida K, et al. Remarkable effect of milnacipran, a serotonin–noradrenalin reuptake inhibitor (SNRI), on depressive symptoms in patients with Parkinson’s disease who have insufficient response to selective serotonin reuptake inhibitors (SSRIs): two case reports. Prog Neuropsychopharmacol Biol Psychiatry. 2005;29(2):351-353.
36. Calvi A, Fischetti I, Verzicco I, et al. Antidepressant drugs effects on blood pressure. Front Cardiovasc Med. 2021;8:704281.
37. Wathra R, Mulsant BH, Thomson L, et al. Hypertension and orthostatic hypotension with venlafaxine treatment in depressed older adults. J Psychopharmacol. 2020;34(10):1112-1118.
38. Taylor C, Fricker AD, Devi LA, Gomes I. Mechanisms of action of antidepressants: from neurotransmitter systems to signaling pathways. Cell Signal. 2005;17(5):549-557.
39. Gillman PK. Tricyclic antidepressant pharmacology and therapeutic drug interactions updated. Br J Pharmacol. 2007;151(6):737-748.
40. Jackson WK, Roose SP, Glassman AH. Cardiovascular toxicity and tricyclic antidepressants. Biomed Pharmacother. 1987;41(7):377-382.
41. Jost WH. A critical appraisal of MAO-B inhibitors in the treatment of Parkinson’s disease. J Neural Transm (Vienna). 2022;129(5-6):723-736.
42. Huang YH, Chen JH, Loh EW, et al. The effect of monoamine oxidase-B inhibitors on the alleviation of depressive symptoms in Parkinson’s disease: meta-analysis of randomized controlled trials. Ther Adv Psychopharmacol. 2021;11:2045125320985993.
43. Finberg JP. Update on the pharmacology of selective inhibitors of MAO-A and MAO-B: focus on modulation of CNS monoamine neurotransmitter release. Pharmacol Ther. 2014;143(2):133-152.
44. Aboukarr A, Giudice M. Interaction between monoamine oxidase B inhibitors and selective serotonin reuptake inhibitors. Can J Hosp Pharm. 2018;71(3):196-207.
45. Panisset M, Chen JJ, Rhyee SH, et al. Serotonin toxicity association with concomitant antidepressants and rasagiline treatment: retrospective study (STACCATO). Pharmacotherapy. 2014;34(12):1250-1258.
46. Smith KM, Eyal E, Weintraub D. Combined rasagiline and antidepressant use in Parkinson disease in the ADAGIO study: effects on nonmotor symptoms and tolerability. JAMA Neurol. 2015;72(1):88-95.
47. Richard IH, Kurlan R, Tanner C, et al. Serotonin syndrome and the combined use of deprenyl and an antidepressant in Parkinson’s disease. Parkinson Study Group. Neurology. 1997;48(4):1070-1077.
48. Vismara M, Benatti B, Nicolini G, et al. Clinical uses of bupropion in patients with Parkinson’s disease and comorbid depressive or neuropsychiatric symptoms: a scoping review. BMC Neurol. 2022;22(1):169.
49. Remy P, Doder M, Lees A, et al. Depression in Parkinson’s disease: loss of dopamine and noradrenaline innervation in the limbic system. Brain. 2005;128(Pt 6):1314-1322.
50. Kumar S, Kodela S, Detweiler JG, et al. Bupropion-induced psychosis: folklore or a fact? A systematic review of the literature. Gen Hosp Psychiatry. 2011;33(6):612-617.
51. Godschalx-Dekker JA, Siegers HP. Reduction of parkinsonism and psychosis with mirtazapine: a case report. Pharmacopsychiatry. 2014;47(3):81-83.
52. Sotto Mayor J, Pacheco AP, Esperança S, Oliveira e Silva A. Trazodone in the elderly: risk of extrapyramidal acute events. BMJ Case Rep. 2015;2015:bcr2015210726.
53. Sarwar AI. Trazodone and parkinsonism: the link strengthens. Clin Neuropharmacol. 2018;41(3):106-108.
54. deMarcaida JA, Schwid SR, White WB, et al. Effects of tyramine administration in Parkinson’s disease patients treated with selective MAO-B inhibitor rasagiline. Mov Disord. 2006;21(10):1716-1721.
55. First MB, Yousif LH, Clarke DE, et al. DSM-5-TR: overview of what’s new and what’s changed. World Psychiatry. 2022;21(2):218-219.